目的
完全長cDNAライブラリーを作製する方法として開発されたOkayama-Berg法(Okayama and Berg, 1982)は、ベクタープライマーやリンカーDNAの調製など多くの熟練した技術を必要とします。そこで、著者らはOkayama-Berg法の長所を取り入れながらも、より簡便なcDNA合成法の開発を試みました。
方法
pBR322をEcoRV切断後、両端に20〜30個のオリゴ(dC)テールを付加してクローニングベクターを調製します。ポリ(A)+RNAを鋳型にしてオリゴ(dT)プライマーを用いて第一鎖cDNAを合成した後、Okayama-Berg法に倣い、RNase H、大腸菌DNAポリメラーゼ I、大腸菌DNAリガーゼによって第二鎖cDNAを合成します。ついでTdTaseにより、二本鎖cDNAの両端に20〜30個のオリゴ(dG)テールを付加します。オリゴ(dC)テールを有するベクターとオリゴ(dG)テールを有する二本鎖cDNAをアニールしたのち、大腸菌の形質転換を行い、cDNAライブラリーを作製します。
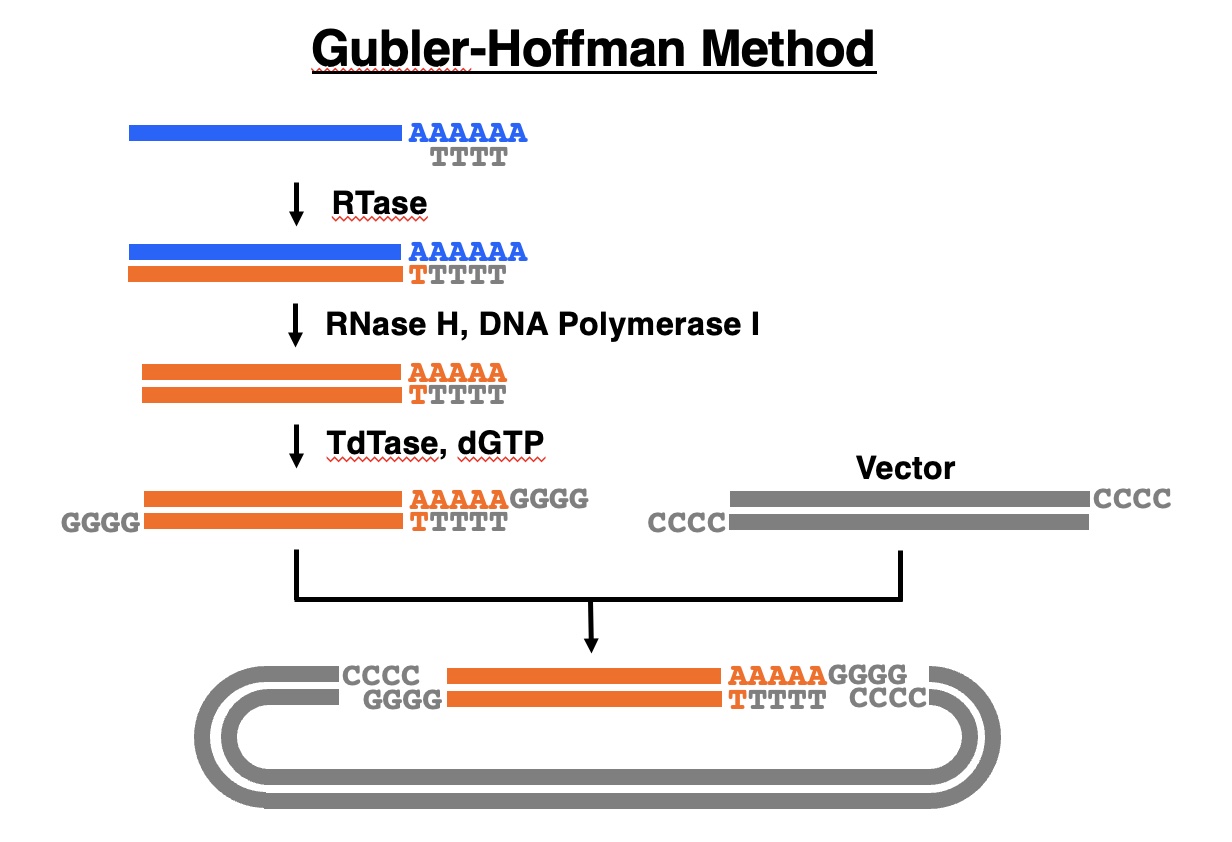
結果
ウサギ網状赤血球のポリ(A)+RNAを鋳型にして、本法により二本鎖cDNAを合成し、その生成物をアルカリアガロースゲルで解析しました。その結果、グロビンmRNAのサイズ(約600塩基)の主バンドとその2倍の長さの副バンドの2本が観測されました。副バンドは、セルフプライミングで生成したものと考えられます。なおこれらのバンドはDNAリガーゼを添加しなくとも生成が認められました。
二本鎖cDNAの両端にオリゴ(dG)テールを付加した後、オリゴ(dC)テールを有するクローニングベクターに挿入して作製したライブラリーから10個のクローンを取り出し、制限酵素解析の結果から5クローンが”full-length”であり、2クローンがβ-グロビンcDNAであることがわかりました。”full-length”と思われるα-グロビンとβ-グロビンcDNAそれぞれ1個の5’端塩基配列を決定したところ、それぞれキャップ部位から15番目と16番目から始まるクローンでした。
本法を用いてウシ副腎髄質からcDNAライブラリーを作製し、プレプロエンケファリン(mRNAのサイズが約1,200塩基)のcDNAをクローン化しました。12クローンのうち8クローンが1kbp以上のcDNAインサートを有しており、2クローンが開始コドンを含んでいました。この2クローンの5’端塩基配列を決定したところ、1クローンはすでに報告されたものと同じであり、もう1クローンは5’端がこれより48bp長いcDNAでした。なお、プライマー伸長法によるとすでに報告されたものより65塩基長いと報告されているので、得られたcDNAはキャップ部位から始まる完全長cDNAではありません。
評価
本論文の最も重要な点は、第一鎖cDNA:mRNA複合体にRNase Hと大腸菌DNAポリメラーゼ Iを作用させると、第一鎖cDNAとほぼ同じサイズの第二鎖cDNAが合成されることを見出したことです。しかもこの際DNAリガーゼは不必要であることが示されました。Okayamaらが想定した第二鎖cDNA合成のメカニズムは、RNase HによってmRNAにニックが入り、DNAポリメラーゼ IがこのニックからRNAをプライマーにしてDNA鎖を合成し、生成したDNA断片をDNAリガーゼで連結するというものでした(Okayama and Berg, 1982)。もしこのメカニズムが正しければ、DNAリガーゼを添加しない場合、第二鎖cDNAは短いDNA断片になるはずです。しかしほぼmRNAと同じサイズの第二鎖cDNAが主に生成するということは、mRNAの5’端に近いニックから始まる第二鎖cDNAが主生成物であることを意味し、DNAリガーゼは第二鎖cDNA合成に寄与していないということになります。ただし、長鎖mRNAを鋳型にする場合には、DNAリガーゼの効果が出てくるかもしれません。
完全長cDNA合成の観点からすると、本法には問題が残ります。著者らが得たグロビンcDNAはいずれも5’端が十数塩基欠失したもので、キャップ部位から始まる完全長cDNAではありません。著者らはこれも”full-length”としていますが、開始コドンから始まる翻訳領域を含むものを”full-length”と呼んでいるようです。もし、第一鎖cDNAの3’端がキャップ部位まで到達しているのなら、これにオリゴ(dG)テールを付加すれば完全長cDNAがとれるはずです。しかしキャップ部位から始まる完全長cDNAは得られていないので、第二鎖cDNA合成の工程で、第一鎖cDNAの3’端がすでに欠失していることを示唆しています。
この点に関しては、次のように説明できます。ニックが入ったmRNAの上流部分がプライマーになってcDNAが合成されるので、5’端側のmRNAはcDNAに置き換わらず残ることになります。残ったmRNAもRNase Hあるいは大腸菌DNAポリメラーゼ Iの内在性RNase Hによってさらに分解除去され、大腸菌DNAポリメラーゼ Iの3’→5’エクソヌクレアーゼ活性により第一鎖cDNAが3’端から分解されると考えられます。ただ、dNTP存在下の反応なので、ポリメラーゼ活性により二本鎖の部分は修復され、結果的に平滑末端となると考えられます。一般的にGubler-Hoffman法として用いられているプロトコルでは、第二鎖cDNA合成後、T4 DNAポリメラーゼにより平滑末端化する工程が入っています。
以上の理由で、本法によっては原理的にキャップ部位から始まる完全長cDNAを合成できないことになります。ただたった2工程で完全長に近いcDNAを合成できること、またその後dGテールの代わりにリンカーやアダプターを付加する方法が取られるようになったこと、ベクタープライマーやリンカーDNAなど特別な道具を必要としないことなどから、本法はcDNAライブラリー作製法としてもっとも広く使われるようになりました。翻訳領域を含んでさえいれば良いのであれば、十分実用的な方法であるといえます。
余談
1993年のCurrent Contents No.20の掲載記事で、引用数が多い論文の著者が論文について語る”This weeks citation classics”に、本論文が取り上げられていました。その中で、Gubler氏が本法を開発する至った経緯を語っています。それによるとプロエンケファリンのcDNAをクローン化するためにS1ヌクレアーゼ法を用いたが短鎖cDNAしか得られず、当時発表されたばかりのOkayama-Berg法を試みたがうまく行かなかったこと、Okayama-Berg法では第二鎖cDNA合成時の酵素の割合が鍵を握っているという噂を聞いて、さまざまな条件で第二鎖cDNA合成を行なっているうちに、本論文で示したように、mRNAとほぼ同じサイズの第二鎖cDNAが合成できることを見つけたことを記載しています。最後に、 “I have no problem admitting that the real reason for developing the procedure was my own inability to get the Okayama and Berg method to work successfully. “ と結んでいます。
この論文が発表された1983年、私はNIHの山田吉彦博士の研究室に客員研究員として滞在し、細胞外マトリックスタンパク質のcDNAクローニングを目指していました。そこでOkayama-Berg法を用いてcDNAライブラリーの作製を試みましたが、Gubler氏同様うまくいきませんでした。そんな折、山田先生がこんな方法があるよと言って持ってこられたのがこの論文でした。そこでベクタープライマーを用いて第一鎖cDNAを合成したのち、RNase H、大腸菌DNAポリメラーゼ I、大腸菌DNAリガーゼによって第二鎖cDNAを合成し、これにEcoRIリンカーをつけてからセルフライゲーションを行うことによってライブラリーを作製してみました。この中からマウスラミニンB1鎖の5.3kbpのcDNAをクローン化できました(K87-1)。ただ、運悪く完全長cDNAの5’端から570bpのところにEcoRI部位があったため、5’側が欠失してしまいました。この部分はプライマー伸長によって作製したcDNAライブラリーから取得しています。この論文の中では、cDNA合成法の詳細については記載せず、Okayama-Berg法の変法を用いたと記載しています。