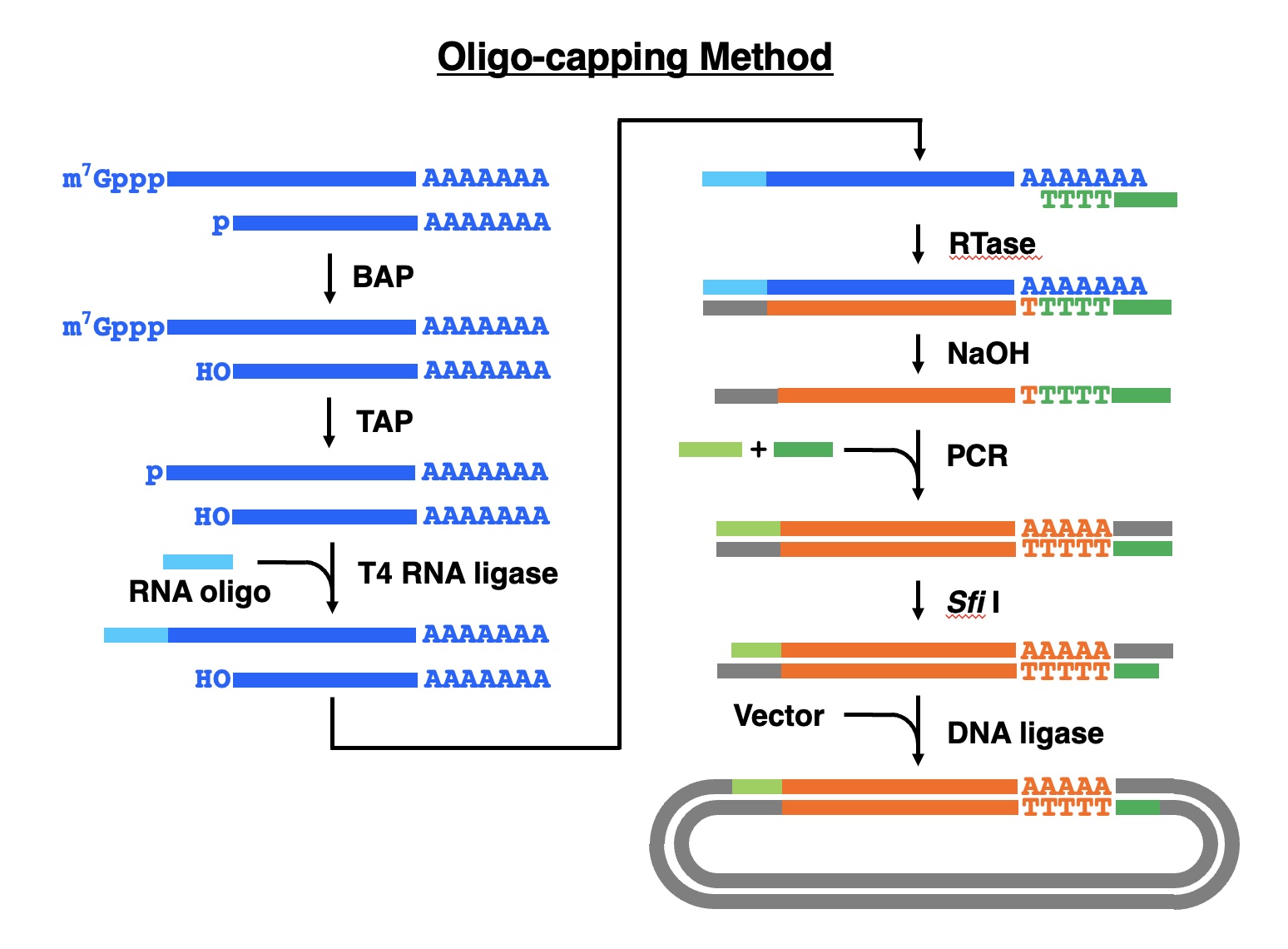
オリゴキャッピング法とは
Okayama-Berg法など従来の完全長cDNA合成法でcDNAを合成した場合、mRNAの5’端まで伸びた第一鎖cDNAを完全長と見なしてきました。しかし、出発材料のmRNAは完全であるとは限らず、分解して短くなったものも含まれています。従って、得られたcDNAが完全なmRNAに由来するかどうかを判別できません。この問題を解決するために、完全なmRNAを選別してからcDNAを合成する方法として考案されたのが、丸山と菅野によるオリゴキャッピング法です。
彼らが着目したのは、mRNAの5’端に存在するキャップ構造です。このキャップ構造を外し、オリゴリボヌクレオチドで置き換えたのちに第一鎖cDNAを合成すれば、キャップ構造を有するmRNAの5’端にのみオリゴリボヌクレオチド由来の配列を有するcDNAを得ることができます。この際、キャップ構造を持たず、5’端にリン酸基を有する分解産物mRNAから前もってリン酸基を外しておけば、不完全なmRNAにはオリゴリボヌクレオチドの連結は起こりません。丸山と菅野はこの方法を「オリゴキャッピング法」と名づけ、1994年に報告しました(Maruyama and Sugano, 1994)。鈴木らはこのオリゴキャッピング法を用いてヒト神経芽腫細胞株からcDNAライブラリーを作製し、得られたcDNAクローンの5’端部分塩基配列を解析したところ、完全長率80%であることが示されました(Suzuki et al., 1997)。
完全長率
この方法では原理的に完全長率100%となるはずですが、初期に作製したcDNAライブラリーの大規模解析の結果得られた値は約60%〜75%となっています(国際出願WO 01/04286 A1)。完全長でないcDNAが生成する原因として二つ考えられます。一つは、分解産物であるmRNAの5’端からのリン酸基の除去が不完全である場合です。元々の出発材料の品質が悪く、分解したmRNAを多く含む場合は、この可能性が高くなります。もう一つは、mRNAを処理する工程中にmRNAの分解が起こり、その分解産物にオリゴリボヌクレオチドが連結してしまうことです。この問題は、ポリ(A)+RNAの代わりに全RNAを用いることにより、改善が認められています(同上文献)。その後、改良法で作製されたライブラリーの大規模解析の結果によると、完全長率は88%に達すると推定されています(Kimura et al., 2006)。
mRNAのサイズと発現量によるバイアス
完全長cDNAライブラリーの作製において完全長率の次に問題となるのが、mRNAのサイズや発現量のバイアスがどの程度かかるかということです。オリゴキャッピング法の場合、PCRによる増幅工程を含んでいるために、個々の遺伝子に由来するcDNAのクローン数に対して両者のバイアスが大きくかかっていると考えられます。特にサイズの面では、長鎖のcDNAの増幅には限界があり、5kbp以上のサイズのcDNAは得られにくいと考えられます。短い遺伝子についても、アガロースゲル電気泳動によるサイズ分画により、1kbp以下の短鎖cDNAを除外しています。また、発現量の少ない希少遺伝子のmRNAの場合、PCRの増幅過程で増幅バイアスがかかり発現量の多いmRNAよりも増幅されにくく、排除されてしまう可能性があります。
PCR増幅とSfiI切断の問題点
PCR増幅工程で見過ごせないのは、人工的に入る変異の可能性とコピークローンの存在です。cDNAをタンパク質生産の目的に使用する場合には変異が入っている可能性を、また発現プロフィール解析に用いる場合にはコピークローンの存在を考慮する必要があります。
もう一つの問題点は、SfiIによる切断工程です。SfiIは8塩基認識の制限酵素なので、cDNA内部にはごく稀にしか存在しないと考えられます。ただcDNAの中にSfiI部位がある場合には、完全長cDNAはクローン化できません。その良い例を我々はLHX3という転写因子のcDNAクローニング過程で見つけました(3.2.2. LIM Homeobox 3 (LHX3) in Kato, 2013)。LHX3遺伝子のサイズは約2.4kbpとそれほど大きくはありません。ベクターキャッピング法で作製したY79のcDNAライブラリーから転写開始点の異なる8個のLHX3の完全長cDNAクローンを同定しているので、この細胞でLHX3は発現量の多い遺伝子になります。しかし、dbESTを見ると同じY79細胞からオリゴキャッピング法で作製したライブラリーからは、LHX3のcDNAは1個も取れていません。調べてみるとLHX3遺伝子の内部には、SfiI部位が1箇所存在していました。この例のように、発現量が多いのにオリゴキャッピング法で作製したライブラリーに含まれていない遺伝子については、SfiI部位の有無を調べてみる必要があります。
転写開始点決定プロジェクト
いくつかの問題点があるにしても、従来法に比べて高い効率でキャップ部位から始まる5’端cDNAを濃縮できるので、大規模に転写開始点を決定するには、有効な方法であると考えられます。鈴木らは34種類のヒト組織と培養細胞からオリゴキャッピング法でcDNAライブラリーを作製し、無作為に選んだ10万クローンの5’端部分塩基配列を解析し、2,251個の遺伝子の完全長cDNAクローンを選別しました(Suzuki et al., 2001a)。これらの配列をヒトゲノム配列と比較することにより、1,031種類の遺伝子の転写開始点とプロモーター領域の同定に成功しました。さらに、276種類の遺伝子から5,880個の転写開始点が同定され、同じ遺伝子であっても異なる複数の転写開始点から始まることが明らかになりました(Suzuki et al., 2001b)。鈴木らはさらに解析数を増やし、DBTSS (Database of Transcriptional Start Sites)というデータベースを構築して公開しました(Suzuki et al., 2002)。現在、Release 10.1までアップデートされています。その後も、ヒト完全長cDNAの5’端配列の大規模解析が続けられ、ヒト遺伝子の多くが選択的プロモーターを有することを明らかにしました(Kimura et al., 2006)。
我々がヒト網膜由来の細胞株に含まれているGAPDHとACTG1についてベクターキャピング法で得た転写開始点の結果と比較してみると、当時のDBTSSに登録されているそれぞれの遺伝子の転写開始点の分布と頻度パターンがほぼ一致しました(Figure 2 in Oshiawa et al., 2011)。我々の方法で得た転写開始点は、cDNAの5’端に余分なdGが付加していることによって、真のキャップ付加部位であることが証拠づけられています。従って、少なくともこれらの遺伝子に関しては、転写開始点の分布に組織特異性はなく、オリゴキャッピング法で得られた完全長cDNAクローンの数に、転写開始点の違いによるバイアスはかかっていないといえます。
FLJプロジェクト
オリゴキャッピング法で作製された完全長cDNAクローンは、転写開始点を決めるだけでなく、遺伝子構造を決めるのに使えることでその本領が発揮されます。そこで、得られた完全長cDNAの全長配列を決定する”full-length long Japan (FLJ)” コレクションプロジェクトが実施されました(Ota et al., 2004)。61のヒト組織、21の初代培養細胞、16の細胞株からオリゴキャッピング法で完全長cDNAライブラリーを作製し、任意に選んだ1,154,510クローンの5’端部分塩基配列を決定しました。この中からRefSeqに一致しなかった総計21,243個の完全長cDNAクローンを選別し、全長塩基配列を決定しました。その結果、10,897個は新規遺伝子であり、その中の5,416個がタンパク質をコードしていました。残り5,481個はノンコーディングRNA候補であり、その4分の1はスプライシングを受けていることが明らかになりました。
総合評価
キャップ構造をオリゴリボヌクレオチドによって置換するという新しい発想により、完全長率80%以上のcDNAライブラリーを作製する技術を確立したという点は、高く評価できます。ただ、遺伝子のサイズや発現量のバイアスがかかることから、1kbp以下の短鎖遺伝子cDNA、5kbp以上の長鎖遺伝子cDNA、希少遺伝子cDNAなどのクローン化は困難であり、全ての遺伝子の完全長cDNAを取りこぼしなく網羅的に収集するという目的には向きません。
いくつかの問題点があるにしても、確実に完全長cDNAクローンを得ることができるので、転写産物の大規模塩基配列決定が可能になりました。その結果、ヒトゲノムプロジェクトの一環として従来実施されてきたEST解析ではなし得なかった、転写開始点の決定によるプロモーター領域の同定を含む遺伝子構造の大規模決定が実現しました。このことがヒトゲノムプロジェクトの中で果たしたオリゴキャッピング法の最大の功績です。