目的
従来の完全長cDNAライブラリー作製法の問題点として、得られたcDNAクローンがmRNAのキャップ部位から開始したものかどうかを判別できないということがあげられます。著者らは以前、mRNAのキャップ構造をオリゴリボヌクレオチドで置き換える方法を開発しました(Maruyama and Sugano, 1994)。そこでこの方法を用いてキャップ部位を有するmRNAを選別したのち、完全長cDNAライブラリーを作製することを試みました。
方法
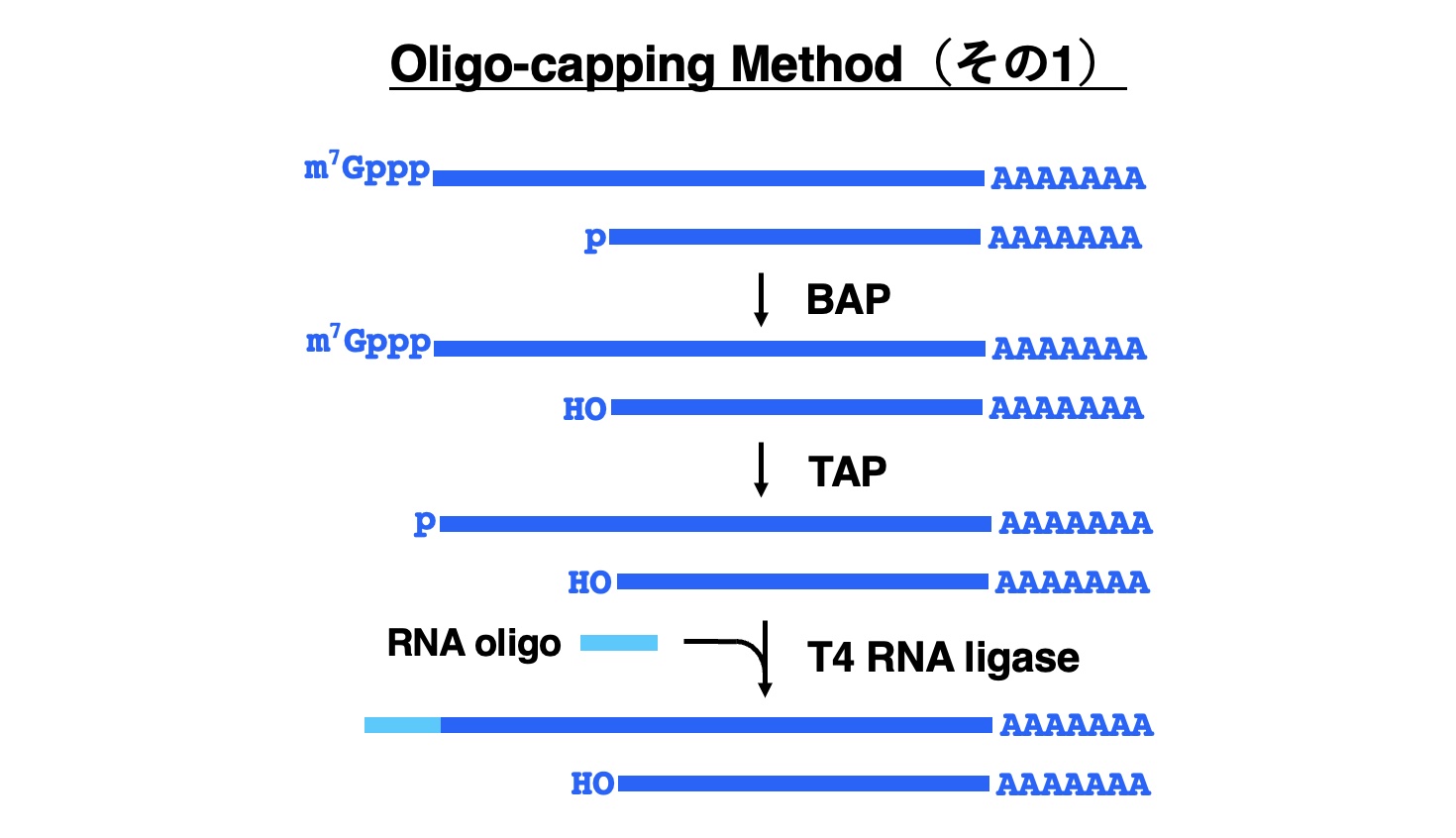
出発材料となるポリ(A)+RNAの中には、キャップのついた完全なポリ(A)+RNAとキャップを持たない分解産物のポリ(A)+RNAが含まれています。そこで最初に、キャップを持たないポリ(A)+RNAの5’端のリン酸基をバクテリア由来アルカリホスファターゼ (BAP) によって除去します。ついでタバコ酸性ピロホスファターゼ (TAP) によってキャップを外し、生成した5’端のリン酸基にT4 RNAリガーゼによってオリゴリボヌクレオチドを連結します。その結果、キャップのついた完全なポリ(A)+RNAの5’端にのみオリゴリボヌクレオチドが連結した産物が得られます。
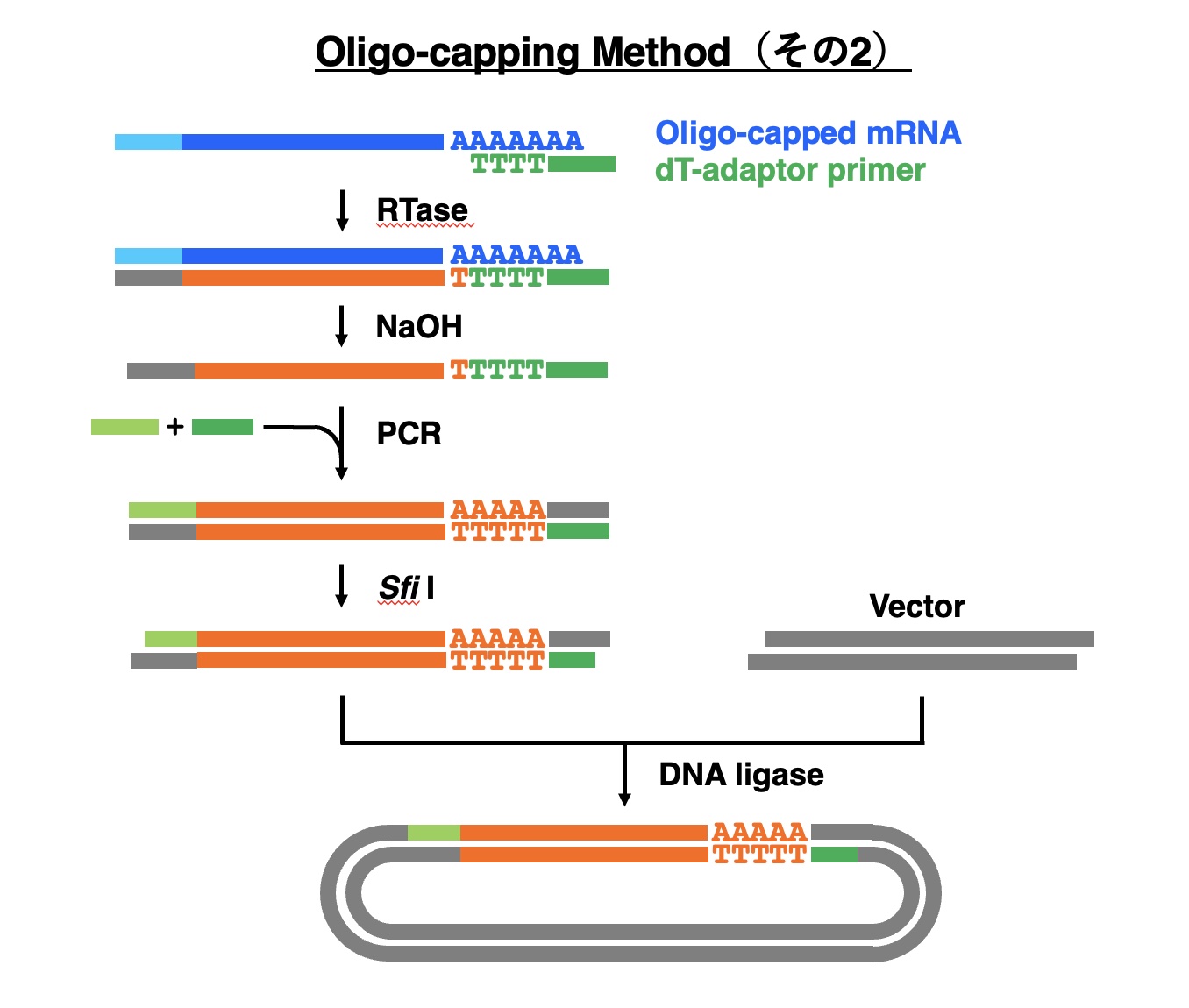
オリゴリボヌクレオチドを連結したポリ(A)+RNAを鋳型にして、dT-アダプターをプライマーにして逆転写酵素(RTase)反応を行い、第一鎖cDNAを合成します。アルカリ処理によってRNAを分解除去したのち、オリゴリボヌクレオチドとdT-アダプターのそれぞれ一部配列を有する2種類のプライマーを用いてPCR反応を行い、cDNAを複製して増幅します。両方のプライマーには異なる配列のSfiI部位が含まれています。PCR産物をSfiI処理した後、アガロースゲル電気泳動にかけてサイズ分画を行い、1kbp以上の長さのcDNAを単離します。得られたcDNA断片をDraIIIで切断したベクターとDNAリガーゼにより連結したのち、大腸菌の形質転換を行い、完全長濃縮cDNAライブラリーを作製します。また、dT-アダプターの代わりに、ランダムアダプタープライマーを用いることにより、キャップ部位を含むポリ(A)+RNAの5’端側を濃縮したcDNAライブラリーを作製します。
結果
ヒト神経芽腫細胞株SK-N-MCから単離したポリ(A)+RNAを鋳型にしてdT-アダプターをプライマーにして完全長濃縮cDNAライブラリーを作製しました。このライブラリーから無作為に選んだ84個のクローンについて、5’端の部分塩基配列を決定したところ、既知遺伝子が35個あり、その中の28個(80%)がキャップ部位から始まる完全長cDNAでした。一方、ランダムアダプタープライマーを用いて作製した5’端濃縮cDNAライブラリーから選んだ159個のクローンには62個の既知遺伝子が含まれており、その中の51個(82%)がキャップ部位から始まるcDNAでした。
長鎖遺伝子cDNAの有無を見てみると、mRNAのサイズが3,000塩基以上の既知遺伝子の5’端塩基配列とほぼ同じクローンが、完全長濃縮cDNAライブラリーからは最長3,317塩基のAhレセプターを含め2個、5’端濃縮cDNAライブラリーからは最長5,427塩基のPDGFレセプターを始め7個得られました。5’端濃縮ライブラリーには、14,279塩基のラットDynein heavy chainと10,300塩基のヒトGiantinの5’端から開始すると思われるクローンが含まれていました。
評価
キャップ部位を有するmRNAを選別したのち、これを鋳型にしてcDNAライブラリーを作製するというアイデアは、完全長cDNAライブラリー作製法に新しい方向を与えるものとして、高く評価できます。ただ方法論としては面白いのですが、実用化する際には多くの問題点があります。
(1)工程数
mRNAの処理からcDNAをベクターへ挿入するまで、8工程もあります。工程数が多ければcDNAの収率が著しく低下するので、出発材料となるmRNAも多くの量を必要とします。
(2)一本鎖mRNAの処理工程
一本鎖mRNAを処理する工程が3工程あり、特にT4 RNAリガーゼによるオリゴヌクレオチドの連結反応は長時間を要するので、その間にmRNAの分解が起こることが懸念されます。
(3)BAP処理
BAP処理が完全でないと、5’端にリン酸基を有する分解したmRNAにもオリゴヌクレオチドが連結します。
(4)PCR
PCRを使用するので、人工的な変異の出現、コピークローンの生成、mRNAのサイズや発現量によるバイアスなどの問題点があります。
(5)SfiI処理
遺伝子内部にSfiI部位が存在すると、cDNAが切断されるため完全長cDNAをクローン化できません。
(6)サイズ分画
アガロースゲル電気泳動でcDNAのサイズ分画を行うので、短鎖完全長cDNAを排除してしまいます。
さまざまな問題点があるにしても、キャップ部位から始まる完全長cDNAを確実に合成できる方法であり、cDNA合成技術における大きな進歩と言えます。ただ、作製したライブラリーには分解産物に由来すると思われる短鎖cDNAも一定割合で含まれているので、転写開始点から始まるcDNAかどうか判別するという問題は未解決のままです。
短鎖cDNAが生成する原因については、著者らも不完全なBAP処理やmRNAの処理工程におけるmRNAの分解に起因すると考察しています。この二つが最大の要因と思われますので、これらの反応の条件を検討することにより、改善が見込めます。例えば、後にポリ(A)+RNAの代わりに全RNAを用いることにより、mRNAの分解が抑えられるということが示されています(国際出願WO 01/04286 A1)。
完全長率80%という値は、翻訳領域を含むcDNAを取得するという目的では満足できる値ですが、PCRによる人工的変異出現の問題を考えると、タンパク質の生産に用いる場合は、注意を要します。また、PCRにより長鎖cDNAを増幅しにくいということもあり、著者らはこのライブラリーを用いて転写開始点を決めるという方向で研究を進め、ヒト遺伝子のプロモーター領域について大規模解析を行い、多様な転写開始点の存在を明らかにしています。(Suzuki et al., 2001a、Suzuki et al., 2001b)。
余談
オリゴキャッピング法のアイデアは、1991年ごろから菅野純夫博士が提唱していました。この話を聞いて、我々もこれを取り入れることができないかと考えて考案したのが、オリゴリボヌクレオチドの代わりに、DNA-RNAキメラオリゴヌクレオチドを用いる「キメラオリゴキャッピング法」です。この方法については1993年の核酸化学シンポジウムで発表し、その短報が公開されています(K93-4)。菅野氏はオリゴリボヌクレオチドを用いる方法を1994年に雑誌Geneに発表しましたが、cDNAライブラリーの解析はだいぶ遅れて1997年の本論文となりました。本論文は、菅野氏の指導のもと鈴木穣氏が大学院博士課程でおやりになった研究が中心となっています。
我々はキメラオリゴキャッピング法を用いて作製したcDNAライブラリーの解析結果を1994年にGeneに発表しました(K94-4)。菅野グループより早く結果を出すことができたのは、当時我々のグループの方が人員体制が整っていたことに起因します。その後、菅野グループも体制を整え、オリゴキャッピング法を用いたヒト完全長cDNAプロジェクトが(株)ヘリックス研究所を中心に実施され、”full-length long Japan (FLJ)” コレクションが構築されました(Ota et al., 2004)。その成果は、ヒトの主要遺伝子のトランスクリプトーム解明に大きな貢献をしています。