キャップトラッパー法とは
理化学研究所(理研)の林崎グループは、マウスの完全長cDNAクローンを揃えるプロジェクト(理研マウス遺伝子百科事典プロジェクト)を計画し、そのために必要となる完全長cDNAライブラリーの作製法として開発したのが、キャップトラッパー法 (CAP Trapper Method)です。
当時、完全長cDNAライブラリーの作製法として、菅野らのオリゴキャッピング法、我々のキメラオリゴキャッピング法、クローンテク社のCapFinder法(後にSMART法に改名)が注目を集めていました。いずれの方法もmRNAの5’端にあるキャップ構造に着目したものです。
Carninciらは、RNAリガーゼを用いるオリゴキャッピング法やキメラオリゴキャッピング法には、RNAリガーゼ反応の鋳型RNA配列依存性の問題が、またオリゴキャッピング法とCapFinder法には、PCRによる増幅工程による変異の生成や塩基配列によるバイアスの問題があることを指摘しました。これらの問題を解決する手段として、mRNAのキャップ構造をビオチン化することによって、ストレプトアビジンビーズで第一鎖cDNA:mRNA複合体を選別する方法を考案し、「キャップトラッパー法」と名付けました。
1996年に出された最初の論文では、mRNAをビオチン化してから第一鎖cDNAを合成しましたが、長時間のビオチン化反応工程でmRNAの分解が起こる可能性があるので、次の論文(Carninci et al., 1997)では第一鎖cDNAを合成してからビオチン化する改良法を報告しました。その結果、アルカリアガロースゲル上では、より長いcDNAの生成が認められました。
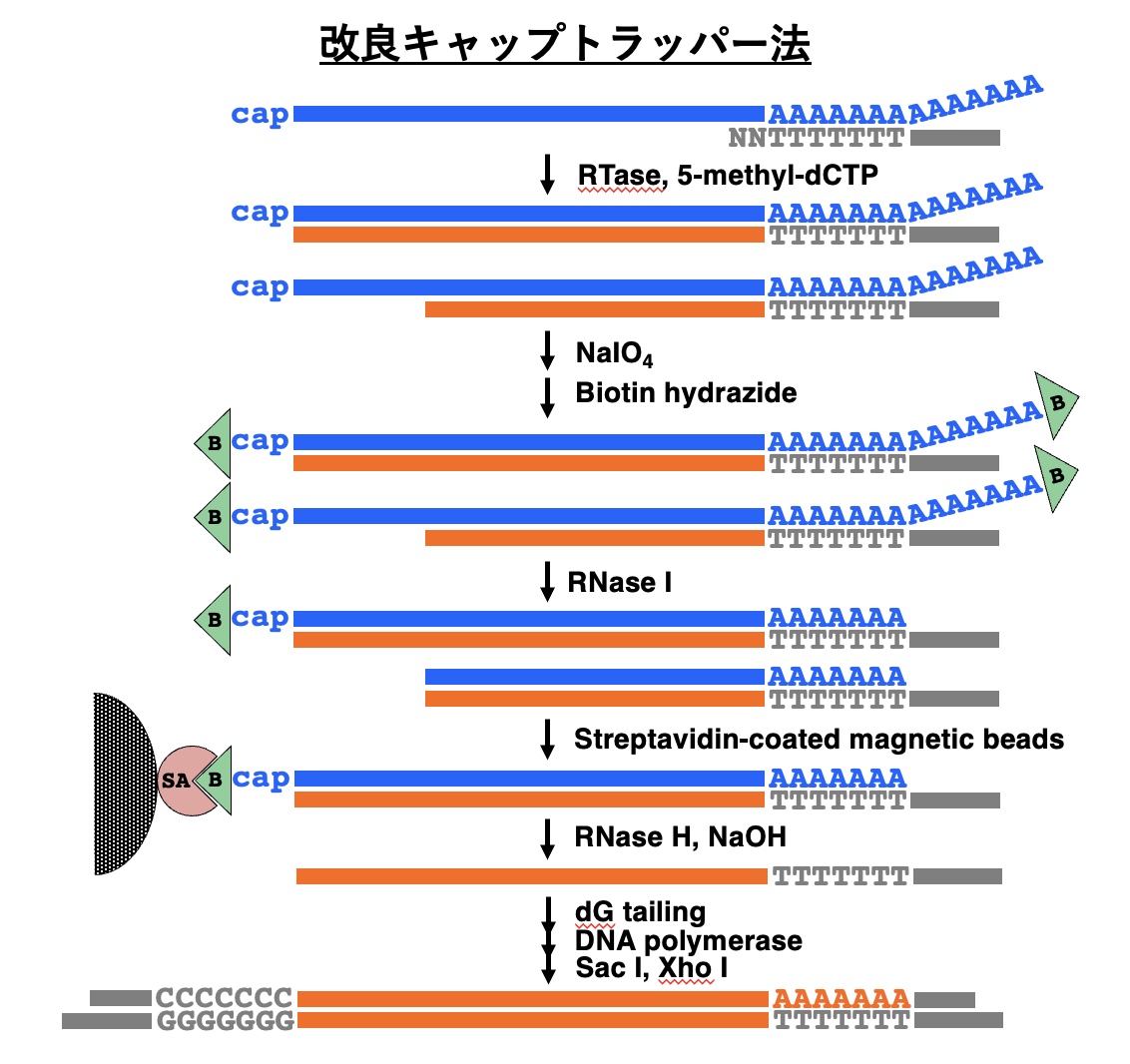
キャップ依存性dG付加の有無
我々は、逆転写酵素(RTase)の末端デオキシヌクレオチジル転移酵素 (TdTase) 活性のため、完全長cDNAの5’端にキャップ依存性のdG付加が起こることを見出しました (K04-1、K05-1)。そこで、キャップトラッパー法で作製した場合、同様のキャップ依存性のdG付加が起こるかどうかに興味がもたれます。原法では完全長cDNAの5’端に余分な塩基配列の挿入は見られず、改良法の論文では得られたcDNAの5’端の塩基配列を決定していません。ただ、改良法で作製したシロイヌナズナのライブラリーについては、65個のクローンの5’端の塩基配列が記載されており、ほとんど全てのクローンがGやTGから始まっています (Seki et al., 1998)。興味深いのは、完全長でないと判定された4個のクローンはGから始まっていないことです。
原法と改良法の結果の食い違いは、先にmRNAのキャップをビオチン化した場合、ビオチン残基による立体障害のため、第一鎖cDNAの3’端にRTaseのTdTase活性によるキャップ依存性dC付加が起こらないことによって説明できます。改良法では、先に第一鎖cDNAを合成するので、3’端にキャップ依存性dC付加が起こりますが、余分なdC付加が起こってもキャップのビオチン化は阻害されなかったと考えられます。
このことから、キャップトラッパー改良法を用いた場合、オリゴdCテールとcDNAの5’端の間に余分なdGがあるかどうかによって、完全長かどうかの判定ができることになります。シロイヌナズナの論文では、この余分なdGについてCapFinder法の説明文(CLONTECHniques January 1996)を引用して、RTaseによる鋳型に依存しない付加であり、クローニングの人工産物とみなしています。もし、これがキャップ依存性dG付加に起因すると認識していれば、完全長であることの証拠であると考えることができたはずです。いずれにしても、この結果はキャップトラッパー改良法によってキャップ部位から始まる完全長cDNAライブラリーを作製できることを証明していることになります。
オリゴdGテーリングの問題点
第一鎖cDNAの3’端にキャップ依存性dC付加が起こるかどうかを判定できるという点では、オリゴdGテーリングを行ったことは正解でした。しかし、cDNAの5’端に長いdCテールが存在すると、塩基配列を決定する際に大きな障害となります。そこで、オリゴdGテールの代わりに一本鎖リンカーを連結する方法、SSLLM (single-strand linker ligation method)が開発されました(Shibata et al., 2001)。
ただ、この方法にもいくつか問題点があります。一つは4,096種類のリンカーを使用するので複雑な反応系になること、そのためcDNAの5’端の配列が本来のmRNA由来の配列なのか、リンカー由来の配列なのか分からないということです。一本鎖cDNAとリンカーとの反応が一晩と長いので、一本鎖cDNAが分解しその分解産物にリンカーが結合する可能性もあります。また一本鎖cDNAの場合、セルフプライミングが起こったり、二次構造を形成したりして、リンカー結合の際、mRNAの5’端の配列によるバイアスがかかる可能性があります。
RTase活性の向上
Carninciらが完全長cDNA合成における問題点の一つとして挙げたのが、一本鎖であるmRNAの二次構造形成により、RTaseが途中で停止してしまうことです。この問題を解決する方法として、反応温度を上げることが考えられますが、RTase活性の低下が懸念されます。そこで高温でもRTaseの活性を維持する条件の検討を行い、トレハロースの添加が有効であることを見出しました (Carninci et al., 1998)。その後、RTaseによる第一鎖cDNA合成をトレハロースの存在下、段階的に温度を上げていって、最終的に56℃で反応を行っています(Carninci et al., 2000)。ただ、この条件がどの程度ライブラリーの完全長率や複雑度の向上に寄与しているかを示す具体的なデータはありません。
長鎖cDNAを得る工夫
次に問題点として挙げたのは、いかにして長鎖cDNAを得るかということです。オリゴキャッピング法やCapFinder法では、PCRを使うため生成するcDNAのサイズに限界があります。PCRを用いないキメラオリゴキャッピング法とキャップトラッパー法ではこの問題は生じませんが、一本鎖であるmRNAの処理工程で分解が起こる可能性が高く、長鎖cDNA合成が難しい原因の一つになります。キメラオリゴキャッピング法では、これが完全長率の低下や長鎖cDNAが得られない最大の要因と考えられます。しかし、キャップトラッパー法ではこの問題は先に第一鎖cDNAを合成してからビオチン化することよって解決されました。
長鎖cDNAを得る上でもう一つの問題点は、クローニングベクターに挿入可能なインサートサイズに限界があることです。そこで、より長いサイズのインサートを収容可能であり、プラスミドライブラリーにも転換可能なクローニングベクターとして、λ-full-length cDNA (λ-FLC) クローニングベクターが開発されました (Carninci et al., 2001)。このベクターを用いることによって、インサートの平均鎖長の増大が認められました。ただ、どれだけ長いサイズの完全長cDNAが得られたかの具体的な結果の記載はありません。
希少cDNAを得る工夫
最後の問題点は、いかにして希少cDNAを得るかです。この問題を解決するために以前から高含量のcDNAを除去するサブトラクション法が種々試みられてきました。Carninciらは、キャップトラッパー法とビオチン化ドライバーを用いたサブトラクション法を考案し、希少cDNAの含有率を上げることに成功しています (Carninci et al., 2000)。ただ、出発材料とし5~10μgのmRNAが必要であり、多量の材料が得られない組織からcDNAライブラリーを作製するのは困難であるという問題は残ります。
FANTOM (Functional Annotation of The Mammalian Genome)の評価
理研は上記の技術を用いて、マウスの各組織から完全長cDNAライブラリーを作製し、5’端と3’端の大規模塩基配列決定により、マウストランスクリプトーム解析を行いました(Okazaki et al., 2002)。世界中から研究者を集めたFANTOMコンソーシアムによって、60,770個の完全長cDNAに機能注釈をつけたとあります。マウスのさまざまな組織から246種類の完全長cDNAライブラリーを作製し、この中から1,442,236クローンの3’端、ついで5’端の塩基配列を決定して選別したcDNAコレクションです。その結果、多くの新しい遺伝子を見つけています。特に、ノンコーディングRNAやアンチセンスRNAが予想外に多かったことは注目に値します。
このコレクションの品質を評価するため、理研のFANTOMのサイトに載っているデータを見てみました。このサイトに記載されているcDNAのサイズ分布の図を見ると、5kbp以上のサイズのクローンは激減しており数えるほどしかありません。7kbp以上の長鎖cDNAが20個登録されているので、各塩基配列を用いて最新のマウス遺伝子データベースでBLAST検索を行ったところ、18個は完全長である可能性が高いですが、残りの2個はキメラクローンでした。完全長と判定した18個のうち、15個がゲノムにはないCGGから始まっています。元となるライブラリーがオリゴdGテール法とSSLL法のどちらの方法で作製されたのかわかりませんが、オリゴdCとキャップ依存性dG付加に由来するものであれば興味深いです。ただ、Gが2〜3個ついていることから、リンカーによるものと思われます。
問題はキメラクローンの存在です。もっとも長い12,349bpのクローン (AK090125)は、驚いたことに4つの遺伝子のキメラでした。1〜2,137がヒト第11染色体、2,135〜3,063がヒト第14染色体、2,069〜11,416がマウス第8染色体、11,417〜12,349がマウス第6染色体に由来します。1〜2,137は、ヒトLMO1の第一エクソンとオーバーラップするアンンチセンス鎖の配列と一致しますが、2,135〜3,063に対応するヒト遺伝子は見つかりません。もっとも長い3番目のマウス第8染色体の配列は、内在性同種志向性マウス白血病ウイルス1 (Mus musculus endogenous ecotropic murine leukemia virus 1)をコードしている領域です。なお、完全長と判定した約8kbpの4個のクローンも、この領域に由来するものです。最後のマウス第6染色体の領域に遺伝子は見つかりません。
もう一つのキメラクローンは、10,481bpのクローン(AK090138)です。このクローンの1~3,063は、上記12,349bpのクローンと全く同じヒト由来配列であり、その下流に完全長と思われるKiaa0100の配列が続いています。両キメラクローンで気になるのは、ヒトの配列とマウスの配列の境界にXhoIサイトがあることです。ベクターにcDNAを挿入する際、XhoI処理しているので、そのこととの関連が疑われます。
両者に見られた1~3,063のヒト由来配列と全く同じ配列で、GenBank NucleotideデータベースのBLAST検索を行ったところ、さらに2個のマウス完全長cDNA、9個のイネ完全長cDNA、2個のダイズcDNAがヒットしてきました。イネ完全長cDNAの一つ (AK287429)について塩基配列をよく見てみると、3,060の位置にXhoIサイト(CTCGAG)が、そしてこれと重なってSacIサイト (GAGCTC)があり、その下流3,096から始まる配列は、確かにイネの第5染色体に存在します。SacIサイトから3,096までの配列は、SSLL法で用いた5’リンカーの配列と一致するので、このキメラクローンはこれらの制限酵素部位で連結生成した人工産物と考えられます。1~3,063のヒト由来配列も、第11染色体と第14染色体のキメラですが、これがどのようにして生成したのかは謎です。
マウス、イネ、ダイズの完全長cDNAライブラリーは同じ実験室で作製されていると思われるので、これらのキメラcDNAは、ライブラリー作製時に、ヒト由来の遺伝子が紛れ込んで生成したと考えられます。制限酵素アダプターを用いてクローニングベクターに挿入する際、このようなキメラが生成することは避けられません。したがって、アノテーションの過程で見つけ出し除外すべきです。まして他の生物由来の遺伝子が混じることになると、データ全体の信頼性を損ねてしまいます。
総合評価
キャップトラッパー法は、キャップまで伸びた完全長cDNAを確実に捕捉できるという点では、他の方法に見られない優れた方法であるといえます。ただ、工程が多すぎるので、出発材料として多量のmRNAを必要とすることから、微量の材料しか手に入らない生物や生体組織からのライブラリー作製には適しません。
遺伝子コレクションという観点から見ると、希少遺伝子や長鎖遺伝子の完全長cDNAの取得が望まれます。理研のグループもここに重点をおいて様々な手法の開発を行なってきましたが、目的を達成しているとは言い難いです。例えば、10kbp以上の長鎖完全長クローンも1個得られていますが、144万クローンの解析で7kbp以上の長鎖完全長cDNAが18個しか採れていません。ちなみに我々がベクターキャッピング法で作製したヒト網膜色素上皮細胞株ARPE-19の完全長cDNAライブラリーからは、約2万クローンの解析により、7kbp以上の長鎖cDNAを48クローン同定しています(K08-1)。
さまざまな生物の遺伝子コレクション作製には向いていませんが、確実にキャップ部位の配列を捉えるという利点を活かせば、多量のサンプルが得られる組織を用いて、含有量の多い転写産物の転写開始点を決定するのには使えます。実際、理研はその方向で研究を進めており、キャップ部位の配列のみを解析するCAGE (Cap Analysis of Gene Expression) 法の開発を行い、遺伝子の転写開始点と発現量解析に応用展開しています(Shiraki et al., 2003)。