目的
本論文は、従来法より優れた完全長cDNAライブラリー作製法を開発することを目的としています。従来法として比較の対象にしたのが、オリゴキャッピング法、我々の開発したキメラオリゴキャッピング法、後にSMART法に改名したCapFinder法(CLONTECHniques, January 1996)、CAPture法(Edery et al., 1995)の4つの方法です。
オリゴキャッピング法とCapFinder法は、PCRによる増幅工程を含むのでそのバイアスがかかり、希少cDNAや長鎖cDNAの取得が困難であるとしています。我々のキメラオリゴキャッピング法はPCR工程を含まないが、オリゴキャッピング法と同様、RNAリガーゼを用いるため、鋳型mRNAの塩基配列や二次構造に起因するバイアスがかかるという問題点を指摘しています。CAPture法は大量のmRNAを必要とするので現実的ではないとしています。従来法が有するこれらの問題点を解決した新しい方法を提案しています。
方法
本法の基本原理としては、CAPture法の考え方を踏襲しています。CAPture法は、キャップ結合タンパク質を用いて、キャップまで伸長したcDNA:mRNA複合体を選別して完全長cDNAのみを得ようとする方法ですが、著者らはキャップ構造をビオチンで化学修飾することによって、同様のことを行おうと考えました。キャップトラッパー法の全行程を下図に示します。
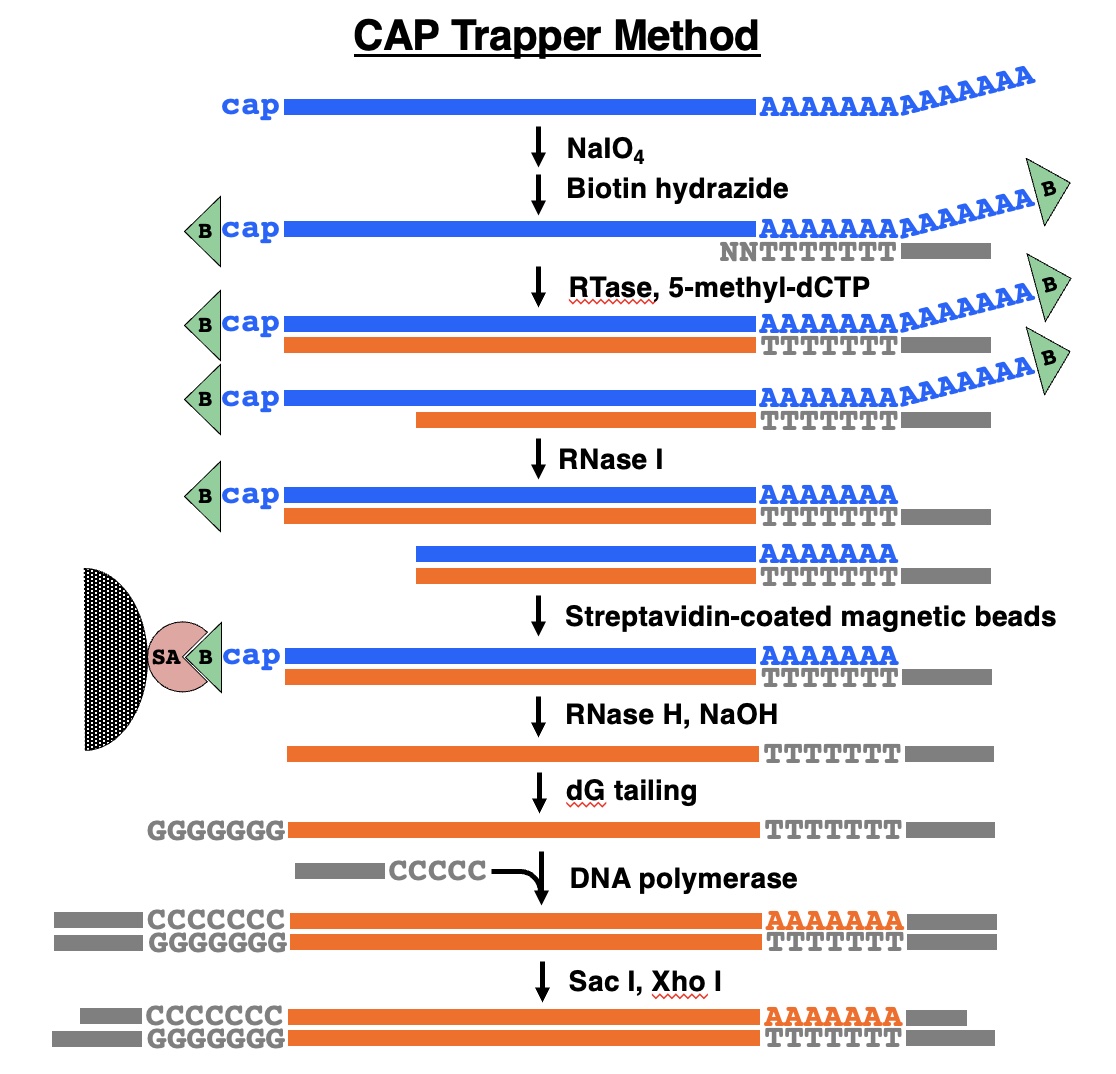
mRNAの5’端にあるキャップ構造のリボースは、隣接した2個の水酸基(ジオール)を有しています。過ヨウ素酸ナトリウムを反応させるとジオールが酸化されて開裂し、ここにビオチンヒドラジドを反応させて、キャップ構造にビオチン基を導入します。mRNAのポリ(A)テールの3’端のAもジオールを有しているので、3’端にもビオチン基が入ることになります。
ビオチン化したmRNAを鋳型として、オリゴdTプライマーアダプターをアニールしたのち、逆転写酵素(RTase)により第一鎖cDNAを合成します。この際、基質としてdCTPの代わりに5-メチルdCTPを用い、後で行う制限酵素処理によってcDNAが切断されるのを防ぎます。なお、オリゴdTプライマーアダプターは5’端にXhoI 部位を有し、12個のオリゴdTに続く3’端の2塩基は縮重配列をとっています。この3’端縮重プライマーを用いることにより、mRNAの長いポリ(A)テールの3’側は末端のビオチン化されたAと共に残ることになります。
第一鎖cDNA:mRNA複合体をRNase I 処理することによって、cDNAとハイブリッドを形成していないmRNAを分解します。その結果、キャップまで伸長していない第一鎖cDNA:mRNA複合体からは、ビオチン化キャップを含む5’側のmRNAとビオチン化Aを含む3’側のポリ(A)テールが分解除去されます
この反応液にストレプトアビジンを固定化した磁気ビーズを添加し、ビオチン化キャップを有する完全長第一鎖cDNA:mRNA複合体をストレプトアビジンに結合して回収します。ついでRNase Hとアルカリ処理によりmRNAを分解すると、完全長第一鎖cDNAが得られます。
その後、完全長第一鎖cDNAの3’端にオリゴdGテールを付加し、SacIとオリゴdCを有する5’側プライマーアダプターを用いてDNAポリメラーゼにより第二鎖cDNAを合成します。ついでSacIとXhoIで消化した後、λZapIIベクターに挿入しcDNAライブラリーを作製します。
結果
マウスの脳から調製した10~20μgのmRNAを用いて、1.2 x 107プラークからなるライブラリーを作製しました。90%以上のプラークはcDNAを有しており、この中から無作為に選んだ983個のクローンについてインサートサイズを調べた結果、平均鎖長は1.6kbpであり、7kbp以上のものも2クローン取れています。GAPDHとEF-1-αについて、5’端と3’端のプローブを用いてプラークハイブリダイゼーションを行った結果、それぞれ98.5%と95%という完全長率が得られました。さらに5’端の塩基配列を決定した結果、データベースに完全長として登録されている5’端配列より長いものがほとんどでした。また120クローンの5’端の塩基配列とデータベースとの比較から、約94%が完全長であると判定しました。
評価
論文に記載された結果を見る限り、完全長cDNAを高い効率で取得するという目的は達成していると思われます。その最大の要因は、キャップ構造を利用して完全長cDNAのみを選別できたことにあります。ただ、トランスクリプトームの大規模解析用ライブラリーという観点から見た場合、次のような多くの問題点があります。
(1)工程数
最大の難点は、cDNA合成の工程数が多すぎるという点です。mRNAの処理からベクターへ挿入するまで10工程もあります。工程数が多ければ収率が著しく低下するので、出発材料となるmRNAも多くの量を必要とします。
(2)一本鎖mRNAの処理工程
mRNAのビオチン化反応は室温で一晩行われますが、この工程でmRNAが分解する可能性があり、長鎖cDNAを取得し難いという問題があります。これはオリゴキャッピング法でも問題となり、5’端の欠けた短縮cDNAが得られる最大の原因でした。ただ、キャップトラッパー法では、分解しても分解産物からは完全長cDNAを選別できないので、分解したmRNAに由来する短縮cDNAがライブラリーに含まれる可能性は低いと考えられます。
(3)2個の縮重塩基を有するオリゴdTプライマーアダプターの使用
第一鎖cDNAを合成する際、3’端に2個の縮重塩基を有するオリゴdTプライマーアダプターを使用しますが、プライミング効率の低下や内部プライミングの可能性が懸念されます。
(4)ストレプトアビジンビーズへの非特異的吸着
著者たちも言及している通り、ストレプトアビジンビーズへ非特異的に短縮cDNAが吸着すると完全長率が低下します。
(5)オリゴdGテール
12〜18個というかなり長いオリゴdGが付加するので、5’端の塩基配列を決定する時に困難を伴います。Fig.4に5’端の塩基配列が記載されていますが、Nが目立ちます。また、cDNAをインビトロ翻訳や動物細胞内で発現させる時も障害になりえます。
(6)一本鎖cDNAを鋳型にした第二鎖cDNA合成
一本鎖cDNAは二次構造を作りやすいので第二鎖cDNA合成の妨げになる可能性があり、またセルフプライミングも起こりえます。
(7)制限酵素サイトを用いるベクターへの挿入
cDNA同士の連結が起こり、キメラやコンカテマーなどの人工産物が形成する可能性があります。
著者らは、この後の論文でいくつかの改良を試みています。(2)の問題を解決するために、先に第一鎖cDNAを合成してからビオチン化を行ったり(Carninci et al., 1997)、(5)の問題を解決するために、オリゴdGテールの代わりに一本鎖リンカーを連結する方法(Shibata et al., 2001 )を報告しています。
我々が開発したベクターキャッピング法と比較すると、cDNAの5’端の配列について一つ疑問が出てきます。我々は、逆転写酵素(RTase)の末端デオキシヌクレオチジル転移酵素 (TdTase) 活性のため、完全長cDNAの5’端にキャップ依存性のdG付加が起こることを見出しました(K04-1、K05-1)。しかし、この論文のFig.4にあるEF-1-αの5’端の塩基配列を見ると、プライマー由来のCとcDNAの間にGが認められません。なお、本論文で我々の論文(K94-4)を引用しているようにEF-1-αの5’端は、CTTTTで始まります。GAPDHのクローンは、Gで始まるものがありますが、これは転写開始点のGです。
この結果は次のように解釈できます。キャップがビオチン化されているために、ビオチン残基が立体障害となり、第一鎖cDNAの3’端にRTaseによるキャップ依存性dC付加が起こらなかったのではないかということです。その後の論文(Seki et al., 1998)で、先に第一鎖cDNAを合成してからビオチン化を行った場合、余分なdGが付加していることからも、この解釈が支持されます。
著者たちが有している問題意識の一つは、従来法では長鎖cDNAのクローニングが難しいということです。Fig.2には、6kbp以上のインサートを有するクローンが7個あり、内2個は8kbp近いと記載してあります。しかし、これらのクローンの5’端配列を決定し、コードしている遺伝子の同定と完全長かどうかの判定をしていません。Nが多すぎてコーディング領域まで読み取れなかったか、あるいはデータベースの中に一致する配列がまだ登録されていなかったかのいずれかと思われます。
さまざまな問題点があるにしても、確実に完全長cDNAを捕獲する方法としての意義は認められる論文です。