ことの始まり
遺伝子工学技術が確立する前は、医薬品として有用なタンパク質の研究は、目的とする活性を有するタンパク質の精製から始まりました。ただ微量しか含まれていないタンパク質を精製するには多くの材料と労力・時間を要します。1970年代にレトロウイルスの逆転写酵素を用いてmRNAからcDNAを合成し、タンパク質の部分アミノ酸配列情報をもとに、目的とするタンパク質をコードするcDNAをクローン化することが可能になりました。
このような技術的背景のもとに、我々は医薬品として有用なヒトタンパク質の探索をタンパク質から迫るのではなく、遺伝子から迫ることを考えました。すなわちヒトの全タンパク質をcDNAの形でそろえ、その中から有用なタンパク質を探索しようという計画です(ホモ・プロテインcDNAバンク構想)。当時ちょうど始まろうとしていたヒトゲノムプロジェクトも、ゲノムの全塩基配列決定のみならず全cDNAのカタログ化を進める方向で動き出しました。
なぜ完全長cDNAが必要か
我々の最終目標はタンパク質を手にすることなので、cDNAは開始コドンから始まる翻訳領域(あるいは読み枠、Open reading frame、ORF)を含んでいる必要があります。翻訳領域を確実に含んでいるのは、mRNAのキャップ部位からポリ(A)テールまでの全領域を含む完全長cDNA(Full-length cDNA)です。完全長cDNAでなくとも開始コドンから始まる翻訳領域が含まれていれば良いわけですが、完全長cDNAでないと真の開始コドンであるかどうかわかりません。したがって、完全長cDNAの合成法を確立することが最初の目的となりました。なお、ゲノム上の転写開始点やエクソン-イントロン構造を決めるためにも完全長cDNAの塩基配列情報は不可欠です。
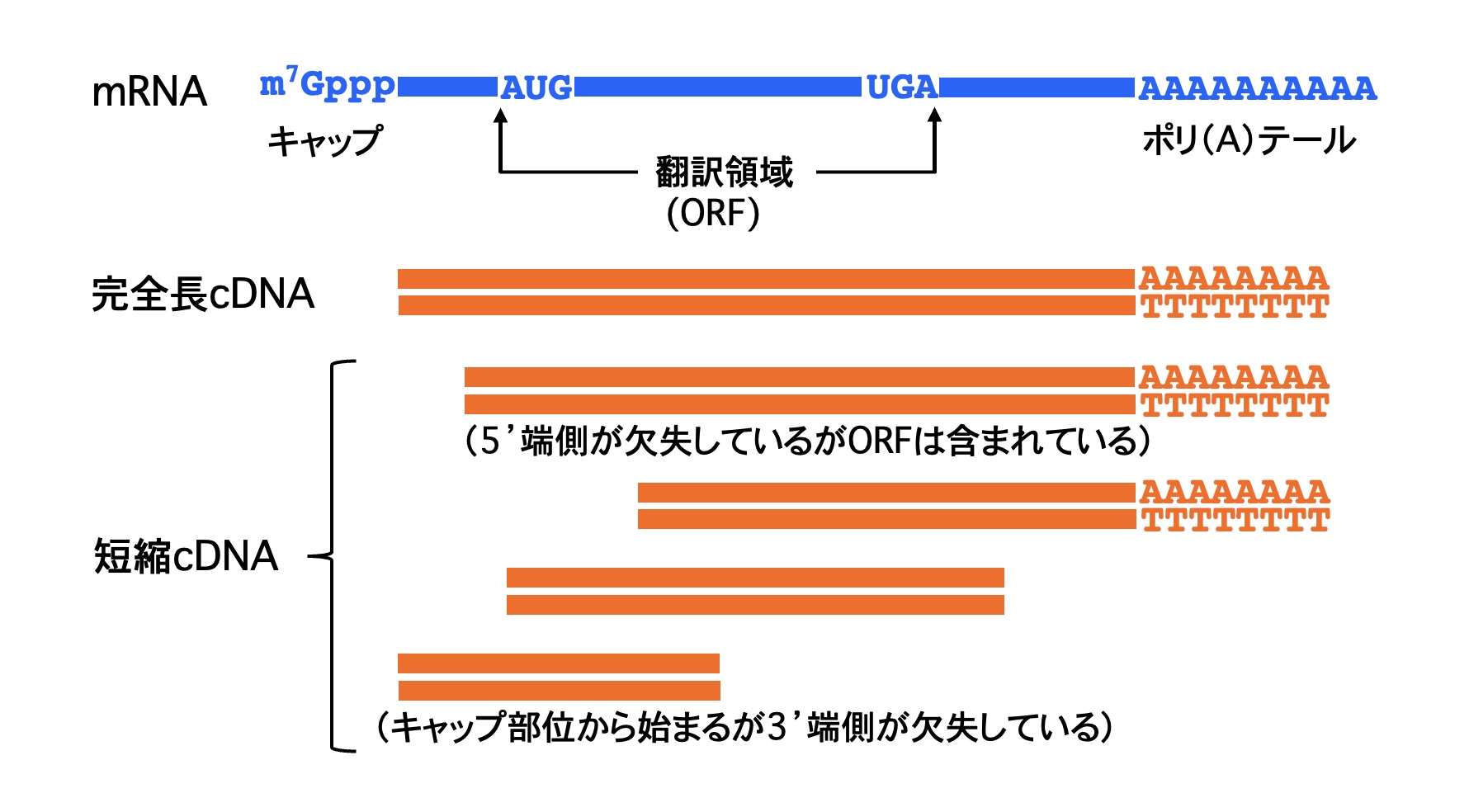
完全長cDNAライブラリーに求められること
完全長cDNAライブラリーの品質に求められるのは、高い完全長率、高い複雑度、人工産物を含まないの3点です。
(1)高い完全長率
高い完全長率を言い換えれば、短縮cDNA (truncated cDNA)を含まないということです。短縮cDNAはmRNAの分解産物から生成するので、分解産物を含まない高品質のmRNAを用いることや、ライブラリー作製工程でmRNAの分解が起こらないようにすることが肝要です。また、生成したcDNAが分解される工程も避ける必要があります。
(2)高い複雑度
一種類のヒト細胞は1万種以上の遺伝子を発現していると考えられています。同じ遺伝子でも転写開始点が異なったり選択的スプライシングなどにより、数種類のmRNA分子種を含んでいます。また、それぞれのmRNA分子種の発現量が異なり、発現量の多い遺伝子と少ない遺伝子でその違いは100倍以上になる場合もあります。さらに、mRNAのサイズも500ヌクレオチド以下から10,000ヌクレオチド以上の広範囲に分布しています。このような複雑度の高いmRNA分子集団に対応した、発現量バイアスやサイズバイアスがかからないライブラリーであることが求められます。特に、発現量が0.01%以下の希少遺伝子cDNAや長さが7kbp以上の長鎖遺伝子cDNAが含まれていることが望まれます。
(3)人工産物を含まない
逆転写酵素やDNAポリメラーゼによる反応で人工的な変異が生成する可能性があります。また、複数の遺伝子のキメラcDNAが生成する可能性もあります。これらの人工産物を含まないことが求められます。
完全長cDNAライブラリー作製法
高品質の完全長cDNAライブラリーに対する上記の要求を満たすために、種々の完全長cDNAライブラリー作製法が開発されてきました。主な方法を下表に示します。なお、この中でS1ヌクレアーゼ法とGubler-Hoffman変法では、完全長cDNAを合成できませんが、比較のため載せてあります。各作製法については「解説と文献」の中のそれぞれの解説や文献に詳しく記載してあります。
| 方法 | 出発材料 | 第一鎖cDNA合成プライマー | 5'端キャップ付加部位保護 | 第二鎖cDNA合成 | 制限酵素処理 | ベクター | 工程数 |
|---|---|---|---|---|---|---|---|
| S1ヌクレアーゼ法 | ポリ(A)+RNA 2~5μg |
オリゴ(dT)プライマー | - | DNAポリメラーゼ | なし | プラスミド | 6 |
| Okayama-Berg法 | ポリ(A)+RNA 2~8μg |
ベクタープライマー | オリゴ(dC)テール | RNase H + DNAポリメラーゼ | (HindIII) | プラスミド | 5 |
| Gubler-Hoffman変法 | ポリ(A)+RNA 3~6μg |
オリゴ(dT)プライマーアダプター | - | RNase H + DNAポリメラーゼ | NotI | プラスミド | 6 |
| Pruitt法 | ポリ(A)+RNA 2~8μg |
ベクタープライマー | オリゴ(dC)テール | RNase H + DNAポリメラーゼ | (BstXI) | ファージミド | 5 |
| オリゴキャッピング法 | ポリ(A)+RNA 5~10μg or 全RNA 5~100μg |
オリゴ(dT)プライマーアダプター | RNAオリゴ | DNAポリメラーゼ (PCR) |
SfiI | プラスミド | 8 |
| キメラオリゴキャッピング法 | ポリ(A)+RNA 10μg |
ベクタープライマー | DNA-RNAオリゴ | RNase H + DNAポリメラーゼ | (EcoRI) | ファージミド | 7 |
| キャップトラッパー法 | ポリ(A)+RNA 1~10μg |
オリゴ(dT)プライマーアダプター | オリゴ(dG)テール or リンカー |
DNAポリメラーゼ | SacI + XhoI | λファージ | 10 |
| SMART法 | ポリ(A)+RNA 1μg or 全RNA 100ng~10μg |
オリゴ(dT)プライマーアダプター | TSオリゴ | DNAポリメラーゼ (PCR) |
SfiI | λファージ | 5 |
| ベクターキャッピンング法 | 全RNA 2~10μg |
ベクタープライマー | ベクター | RNase H + DNAポリメラーゼ | なし or (EcoRI) |
ファージミド | 3 or 4 |
各方法で用いられている出発材料や必要とする道具と基本的な行程を比較してみます。
(1)出発材料RNA
従来法の多くは1~10μgのポリ(A)+RNAを出発材料として用い、ベクターキャッピング法は2~10μgの全RNA (total RNA) を用います。オリゴキャッピング法では5~100μgの全RNAでも、またSMART法では100ngの全RNAでもライブラリー作製可能であることが報告されています。少量の出発材料で済むという点では、全RNAを用いる方法が望ましいです。全RNAを用いる場合、オリゴキャッピング法とSMART法はPCRによる増幅工程を含んでいますが、ベクターキャッピング法はそのような増幅工程を必要としません。
(2)第一鎖cDNA合成
第一鎖cDNA合成はプライマーとしてオリゴ(dT)プライマーを用いるかベクタープライマーを用いるかのいずれかの方法がとられます。ベクタープライマーを用いる場合、ベクタープライマーという特別な道具を作る必要がありますが、最初の工程でmRNAをベクターに組み込んでしまうので、残りの工程で発現量バイアスやサイズバイアスがかかりにくく、mRNAの複雑度を反映したライブラリーが作製できるという利点があります。また、cDNAの向きが決まる、制限酵素によるcDNAの切断が起こらない、キメラcDNAが生成しないといった利点もあります。
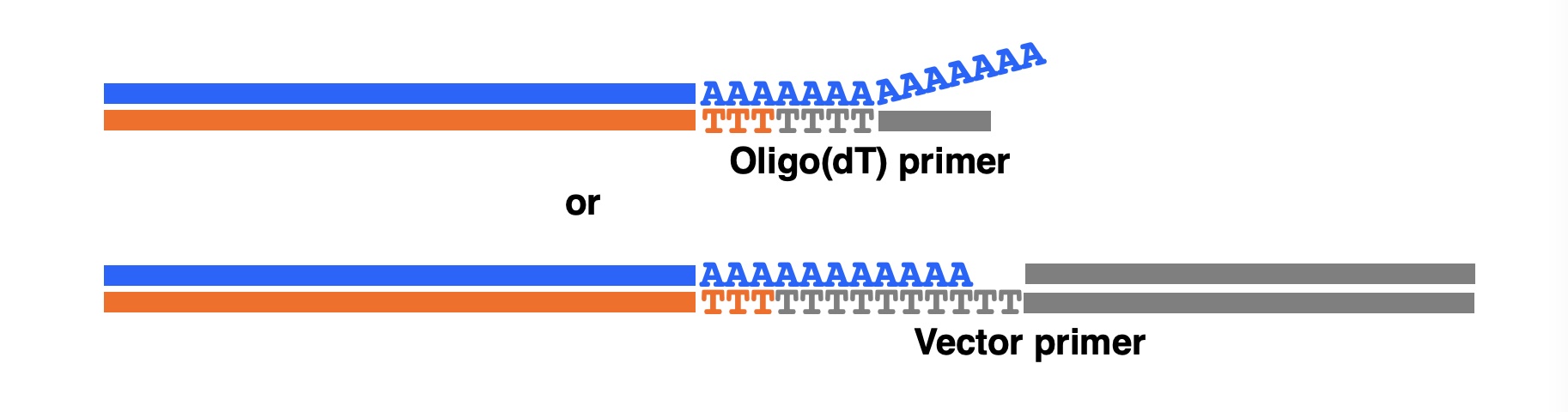
(3)5’端キャップ付加部位の確保
5’端のキャップ構造から開始するcDNAを合成するために、5つの方法がとられます。分解していない完全なmRNAを用いれば、いずれの方法でも5’端が完全なcDNAが得られます。
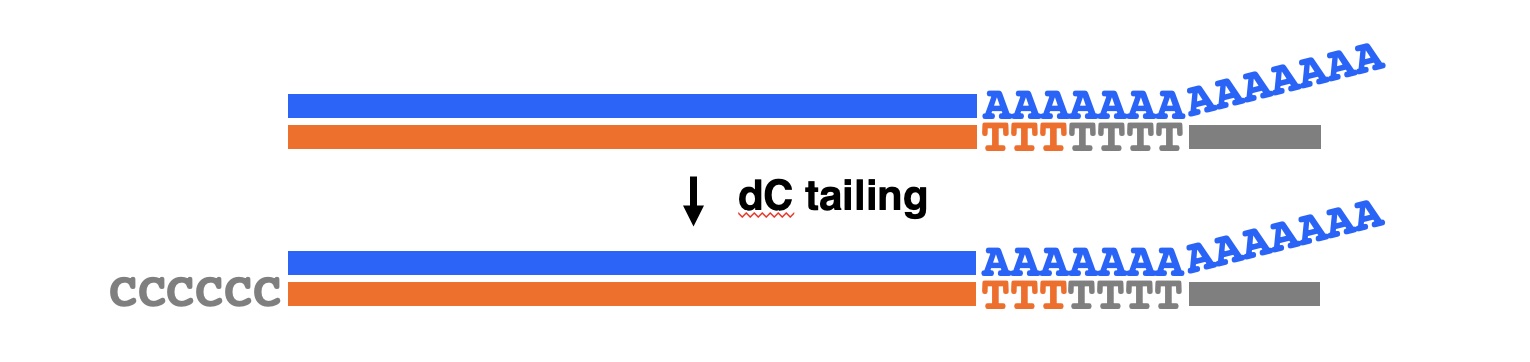
(i)オリゴヌクレオチドテール付加
第一鎖cDNAを合成後、その3’端にオリゴヌクレオチドテールを付加する方法です。Okayama-Berg法、Pruitt法ではオリゴ(dC)テールを、キャップトラッパー法ではオリゴ(dG)テールを付加します。ただこの方法ではテールの数をうまくコントロールするのが難しいという問題点があります。
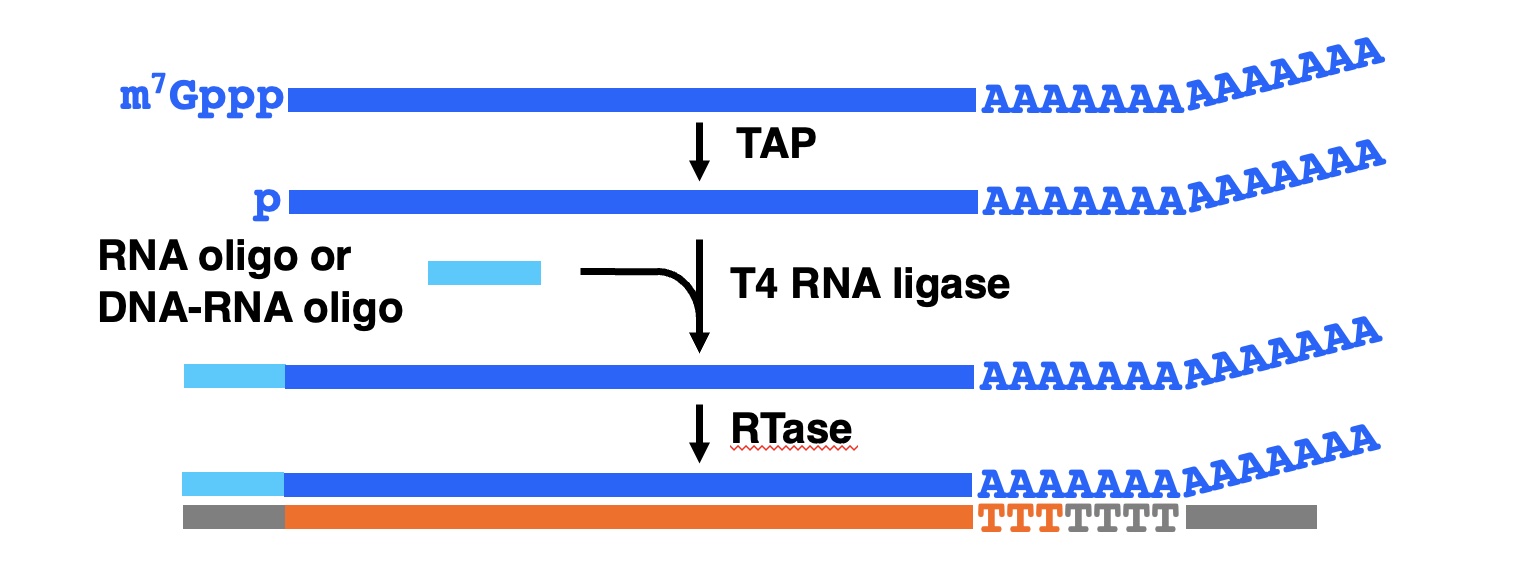
(ii)キャップのオリゴヌクレオチドによる置換(Oligo-capping)
mRNAのキャップ構造をオリゴヌクレオチドで置換したあと第一鎖cDNAを合成する方法です。オリゴヌクレオチドとしてRNAオリゴを用いるのがオリゴキャッピング法、DNA-RNAオリゴを用いるのがキメラオリゴキャッピング法です。
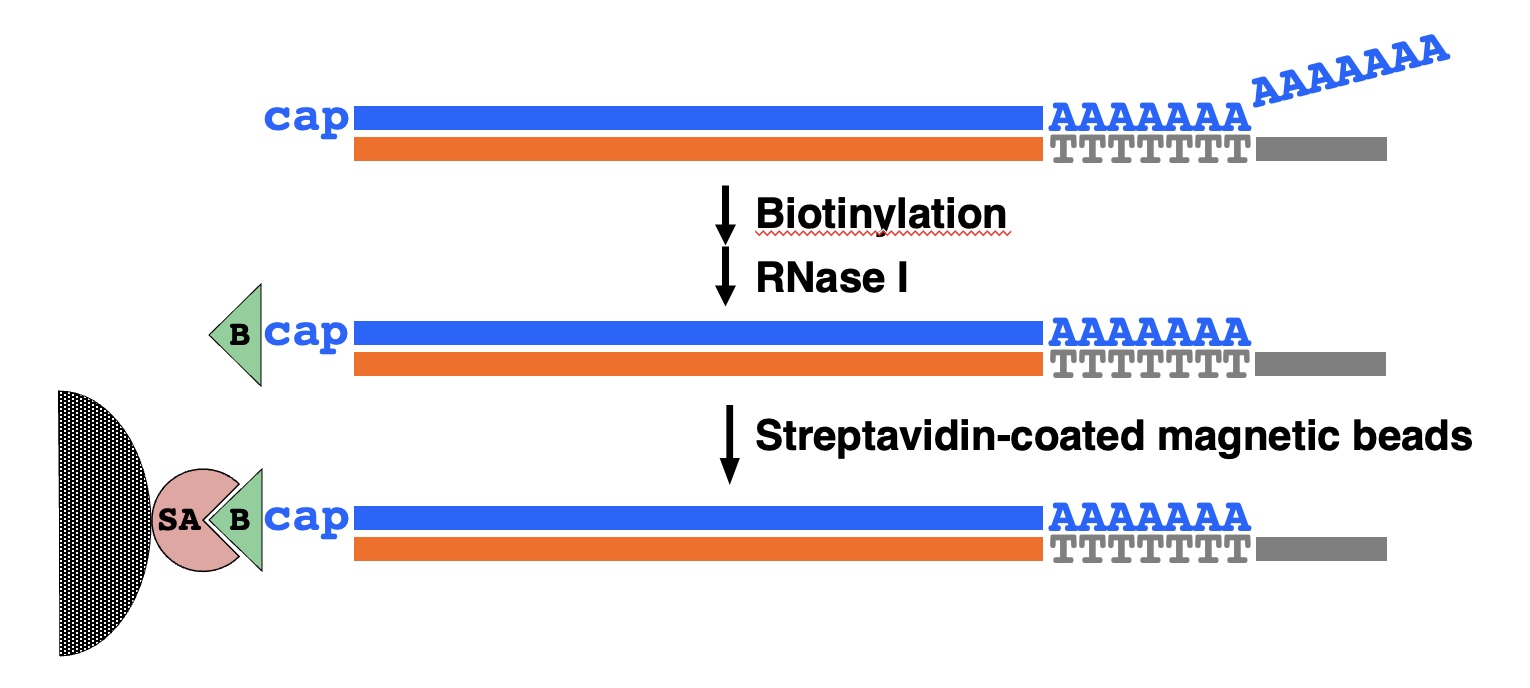
(iii)キャップのビオチン化による選別
mRNAのキャップ構造をビオチン化した後、第一鎖cDNAを合成し、ストレプトアビジンビーズで完全長mRNA:cDNA複合体を選別するのがキャップトラッパー法です。下図に示す改良法では第一鎖cDNAを合成した後、mRNAのキャップ構造をビオチン化しています。
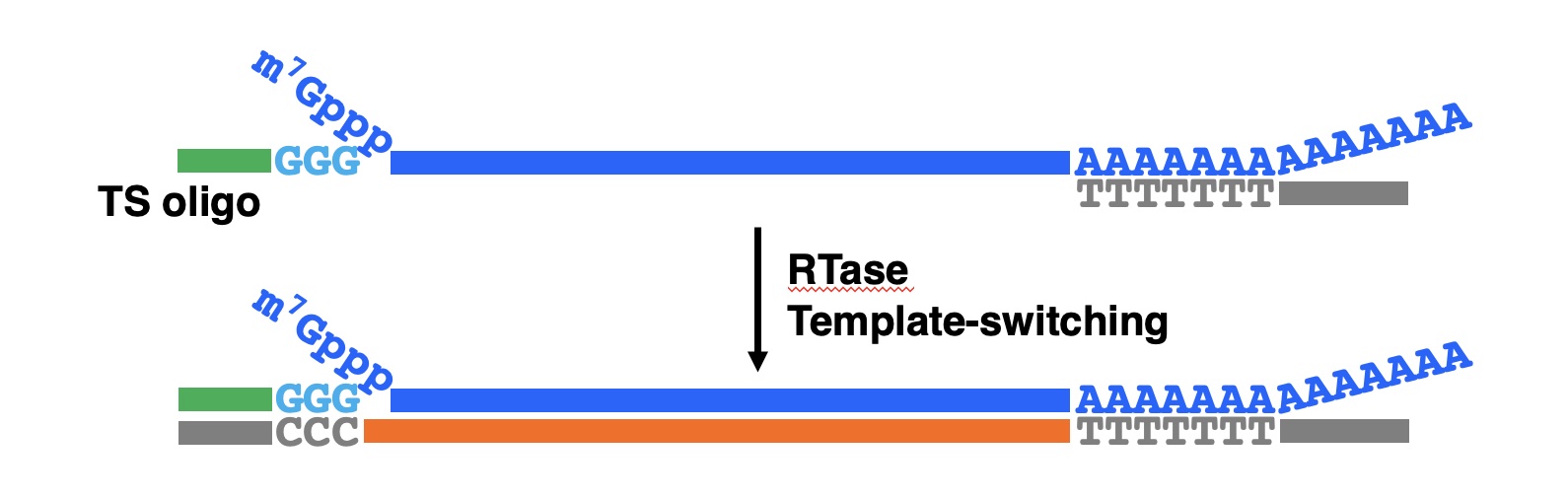
(iv)鋳型交換反応(Template-switching)
逆転写酵素の鋳型交換反応を利用して、第一鎖cDNAの3’端に制限酵素部位を有するオリゴヌクレオチドを付加するのがSMART法です。
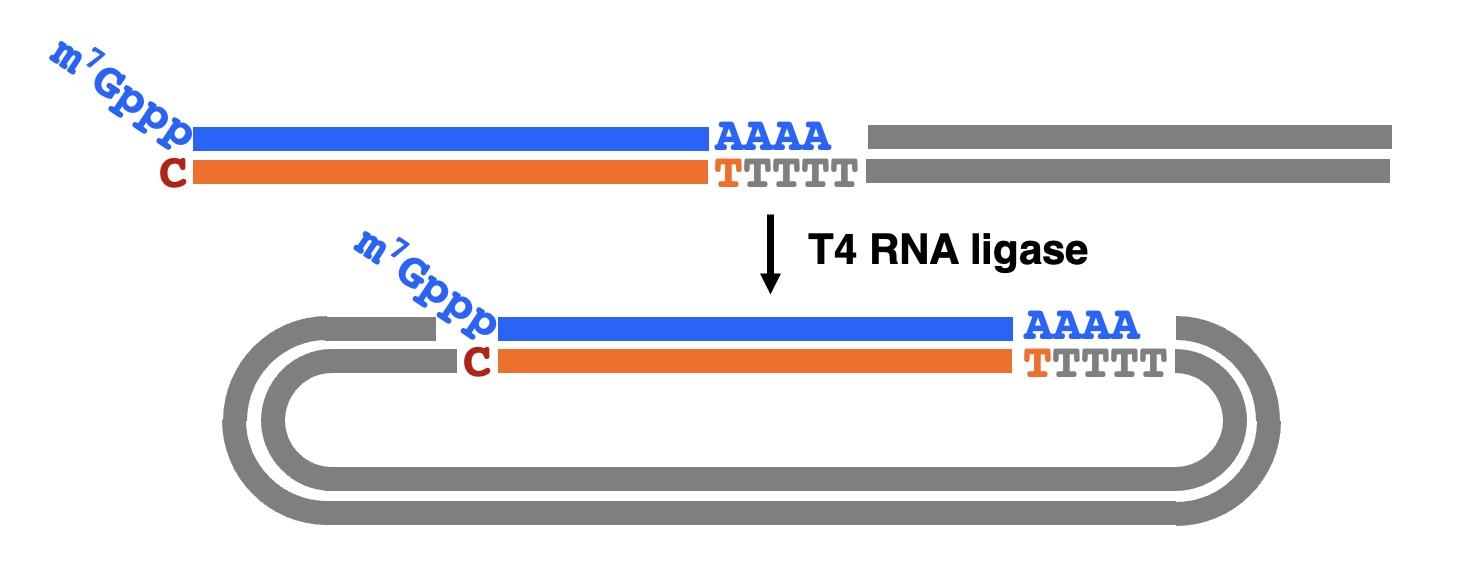
(v)ベクターの直接連結(Vector-capping)
T4 RNAリガーゼによるセルフライゲーションによってmRNA:cDNA複合体の末端にベクターを直接連結するのがベクターキャッピング法です。
(4)第二鎖cDNA合成
第二鎖cDNA合成には、3つの方法がとられています。
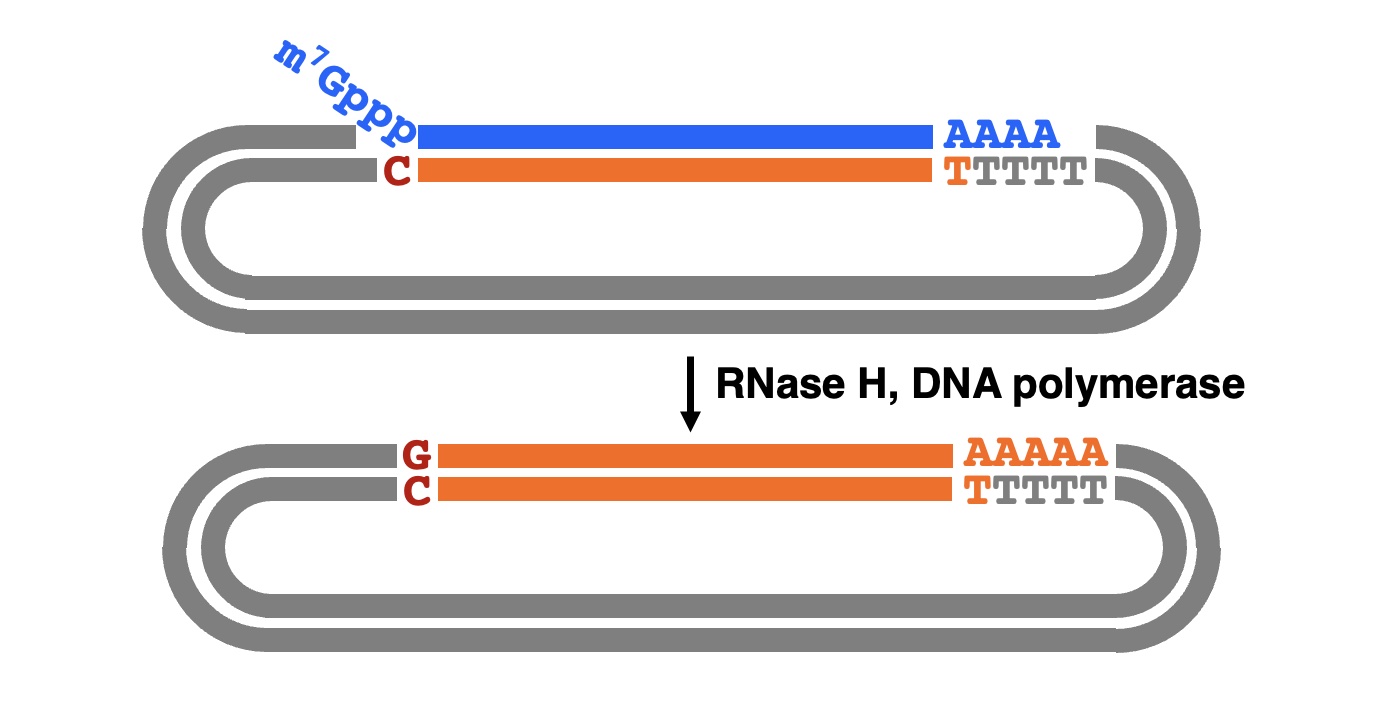
(i)mRNAをDNAで置換
mRNA:cDNA複合体にRNase HとDNAポリメラーゼを反応させてmRNAをDNAで置き換える方法です。この方法では、第一鎖cDNAが物理的に弱い一本鎖の状態になることや二次構造を形成することから避けられます。ベクタープライマーを用いるOkayama-Berg法、Pruitt法、キメラオリゴキャッピング法、ベクターキャッピング法がこれに該当します。
(ii)プライマー伸長反応
第一鎖cDNA合成後、アルカリ処理などにより一本鎖cDNAとしたのち、第一鎖cDNAの3’端に付加したオリゴヌクレオチドにアニールするプライマーを用いて第二鎖cDNAを合成する方法です。キャップトラッパー法がこれに該当します。キャップトラッパー改良法では末端に(dN)6を有するオリゴヌクレオチドリンカーを用いています。
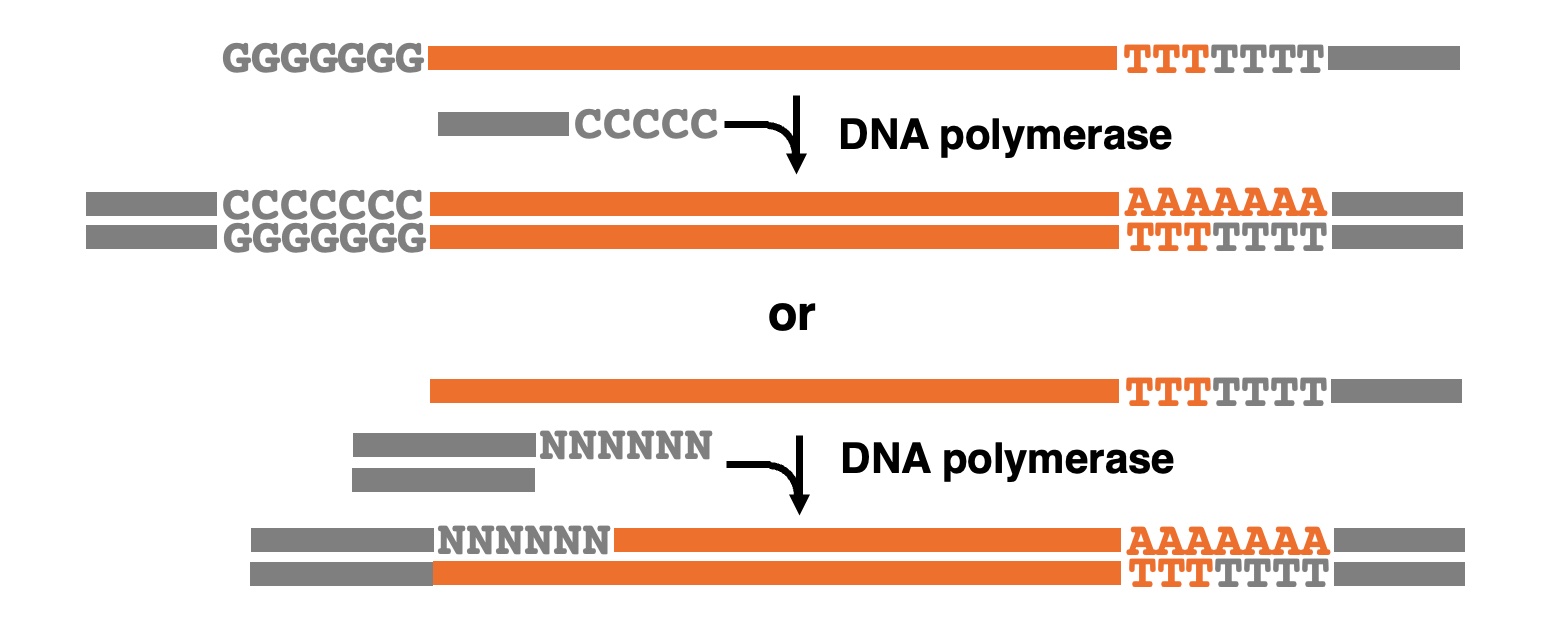
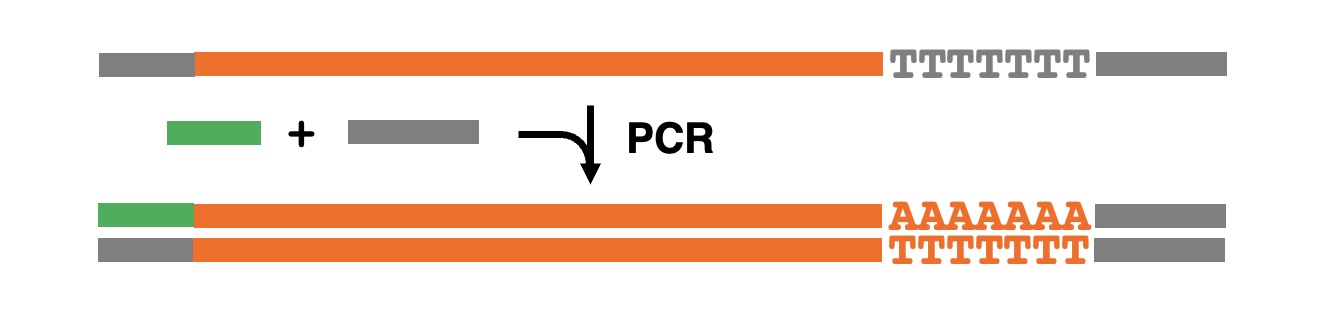
(iii)PCR
一本鎖の第一鎖cDNAを鋳型にして、PCRにより増幅する方法です。オリゴキャッピング法とSMART法がこれに該当します。
(5)制限酵素処理
リンカーやプライマーアダプターを使用する場合、cDNAをベクターに組み込むのに制限酵素切断が必要となります。cDNA内に制限酵素部位があると切断されてしまうので、キャップトラッパー法では第一鎖cDNA合成の際に、基質としてdCTPの代わりに5-メチルdCTPを使用し、cDNA内での切断を防いでいます。またcDNAの両端に制限酵素部位があると、キメラcDNAが生成する可能性があります。これを防ぐために、オリゴキャッピング法とSMART法では、突出末端が非対称の塩基配列を有するSfiIを用いています。ただしこの場合、cDNA内にSfiI部位があると切断されてしまいます。ベクタープライマーを用いる場合でも制限酵素処理工程はありますが、mRNA:cDNA複合体の段階で行うので、cDNA内での切断はほとんど起こりません。
(6)ベクター
ベクターとしてプラスミドとファージミド、あるいはλファージが用いられています。複雑度を反映させるには形質転換効率が高いファージが有利ですが、cDNAの段階で十分な複雑度を確保できなければクローン数が多いだけでは意味がありません。プラスミドやファージミドでも十分形質転換効率が高く106以上のクローンを得ることができるので、その後の解析を考えるとプラスミドやファージミドベクターの方が適しています。なお、プラスミドやファージミドにdTテールを付加したものが、ベクタープライマーとして用いられます。
(7)工程数
表には第一鎖cDNA合成からベクターへ挿入するまでの工程数を示しています。mRNAの処理工程を含むものはそれだけ工程数が多くなります。工程数が多くなると反応中に分解したり収率が低下したりするので、できるだけ少ないことが望まれます。その点、ベクターキャッピング法はたった3~4工程で済むので、他の方法に比べ高い収率や複雑度が期待できます。
完全長cDNAライブラリー作製法の問題点
完全長cDNAライブラリー作製法において、ライブラリーの品質で問題となる点について詳しく考察してみます。
(1)完全長率
完全長率を低下させる要因としては、mRNAの分解、3’端の欠失、5’端の欠失、cDNAの分解があげられます。
(i)RNA抽出・精製時のmRNAの分解
出発材料であるmRNAが完全であることが、高い完全長率を達成するための最低必要条件です。生体組織からRNAを抽出する際、ある程度分解することは避けられません。タンパク質をコードしているポリ(A)+RNAは全RNAの1%~5%程度しか含まれていないので、多くの方法では精製ポリ(A)+RNAを用いていますが、精製過程でのポリ(A)+RNAの分解が懸念されます。オリゴキャッピング法、SMART法、ベクターキャッピング法では全RNAを用いてもライブラリーを作製できることがわかり、その結果、ポリ(A)+RNAを精製する必要がなくなり、出発材料の量を少なく抑えることができるようになりました。
(ii)mRNA修飾工程での分解
生体組織によっては、RNA抽出過程でどうしてもmRNAの分解を免れない場合があります。分解産物を多く含む材料を用いる場合は、オリゴキャッピング法、キメラオリゴキャッピング法、キャップトラッパー法のような完全長cDNAを選別できる方法が有効です。ただし、オリゴキャッピング法やキメラオリゴキャッピング法のように複数の酵素処理工程や長時間の反応を要するmRNA修飾工程を含む場合、この工程で分解した産物が短縮cDNA生成の原因になっていると考えられます。これらの方法も精製ポリ(A)+RNAの代わりに全RNAを用いることによって、完全長率の改善が認められています。キャップトラッパー法も最初はmRNAのキャップ構造のビオチン化工程から始まっていたので、この工程でのmRNAの分解が懸念されましたが、改良法では第一鎖cDNAを合成してからビオチン化しているので、この問題は避けることができています。
(iii)3’端の欠失
第一鎖cDNAを合成するのにオリゴ(dT)プライマーを用いた場合、mRNAの内部にAストレッチがあると、そこからプライミングして3’側が欠失する場合があります。一般にこのようなAストレッチは3’-非翻訳領域(3’-UTR)にあるので、翻訳領域だけが必要である場合には問題ありません。ただ、3’端の2塩基が縮重したオリゴ(dT)プライマーを用いる場合には、翻訳領域内からプライミングする可能性があります。
一方、ベクタープライマーは約60個のdTテールを有するので、短いAストレッチにアニールする可能性は低いと思われます。実際、オリゴ(dT)プライマーを用いて取得したある遺伝子のcDNAが、3’-UTRにあるAストレッチにプライミングして生成したものであることを、我々がキメラオリゴキャッピング法で作製したライブラリーから取得したcDNAと比較して明らかにした経験があります。ただ、ベクタープライマーは短い ポリ(A)テールをもったmRNAを捕捉できない可能性があります。
(iv)5’端の欠失
mRNAの内部で二次構造が生成すると、逆転写酵素の伸長反応がここで停止し、5’端が欠失する可能性があります。表にあげた完全長cDNAライブラリー作製法では、原理的にはこのように途中で伸長反応が停止したものから短縮cDNAは生成しませんが、完全長cDNAも得られないということになります。
反応温度を高くすれば二次構造の形成が抑制されるので、キャップトラッパー法では逆転写酵素の耐熱性を向上させる工夫がなされています。ただ、我々の経験からすると現在市販されてよく使われている逆転写酵素はこのような二次構造があっても逆転写反応が十分進行していると思われます。
他に、一本鎖状態の第一鎖cDNAを用いて第二鎖を合成する場合、セルフプライミングが起こり、完全長cDNAの収率が低下する可能性がありますが、S1ヌクレアーゼ法以外このことによる短縮cDNAの生成は起こらないと思われます。
(v)cDNAの分解
制限酵素処理工程を含む場合、切断防止策を取らないとcDNAの内部で切断される可能性があります。その具体例の一つが、以前、私がOkayama-Berg法の改変法でクローニングしたマウスラミニンB1鎖cDNAです。EcoRIリンカーを用いたのですが、cDNA内部にEcoRI部位があったため、完全長cDNAを得ることができませんでした(こちら)。もう一つの例はヒトのLHX3という遺伝子のcDNAで、内部にNotI、XhoI、SfiIという3つの制限酵素部位があるため、従来法でこれらの制限酵素部位を用いた場合、cDNAが切断されてしまい完全長cDNAが得られていません(こちら)。
(2)複雑度
複雑度を低下させる要因としては、発現量バイアス、サイズバイアス、ライブラリーサイズバイアスが挙げられます。
(i)発現量バイアス
全mRNAに占める個々の遺伝子のmRNAの割合は、1%以上から0.01%以下までと広範囲にわたります。また、発現量が多い遺伝子であっても、複数のスプライシングバリアントが存在し、その中には希少バリンアントが存在することもあります。含有量の少ない希少遺伝子や希少バリアントのcDNAは、ライブラリー作製の各工程で抜け落ちる可能性が高くなります。
その第一の要因は、基質や酵素が含有量の多いmRNAのcDNA合成で消費され、希少mRNAのcDNA合成まで回らないということです。したがって同じ反応条件下では出発材料のmRNA量ができるだけ少ない方が望ましいということになります。その意味でも、精製ポリ(A)+RNAを用いるより全RNAを用いる方が有利です。
希少遺伝子cDNAが抜け落ちる第二の要因は、cDNAの増幅工程におけるバイアスです。例えばPCR工程や大腸菌の形質転換体を増殖する工程を含む場合、コピークローンが生成し高含量のcDNAが優先的に増幅すると考えられます。
このような発現量バイアスを小さくするためには、全RNAを用いる、工程数を少なくする、増幅工程をなくすなどが必要です。また、ベクタープライマーを用いることにより、最初の工程でmRNAをベクターに組み込んでしまうことも、発現量バイアスを小さくするのに有効です。
(ii)サイズバイアス
長鎖遺伝子の完全長cDNAが得られにくい要因としては、RNA調製時のmRNAの分解、mRNA修飾工程時のmRNAの分解、mRNAの二次構造形成による逆転写酵素反応の停止などが挙げられます。また、短いmRNAの方が先にcDNA合成が終了するので、基質や酵素が短いmRNAのcDNA合成で消費さるというように、発現量バイアスと同様の問題が起こりえます。
PCR工程を含む場合は、増幅できるcDNAの長さに限界があります。また、合成されたcDNAをベクターに挿入する工程で長鎖cDNAの挿入効率が低下することや長鎖cDNAの形質転換効率が低下することなどもサイズバイアスとなります。
リンカーやアダプターを用いる場合、未反応のリンカーやアダプターを除去するため、アガロースゲル電気泳動によりサイズ分画を行いますが、この際、短鎖遺伝子cDNAも除去される可能性があります。
サイズバイアスを小さくするには、全RNAを用いる、mRNA修飾工程や増幅工程をなくす、サイズ分画を行わない、長鎖cDNAを挿入できるベクターを選択するなどが要求されます。
(iii)ライブラリーサイズバイアス
十分な形質転換体が得られないと、複雑度を反映したライブラリーにはなりません。形質転換効率だけを考えるなら、ベクターとしてλファージなどを用いた方が良いと思われますが、プラスミドやファージミドでもエレクトロポレーション法を用いることにより、十分な形質転換効率が得られるようになってきました。したがって、cDNA合成時の複雑度を保つことの方が重要です。
(3)人工産物の生成
cDNA合成工程で生成する人工産物は、変異クローン、キメラクローン、コピークローンです。
(i)変異クローン
逆転写酵素やDNAポリメラーゼは、低頻度ですが間違った塩基を取り込んで点変異を生じます。特に、PCRによる増幅工程では複数サイクルのポリメラーゼ反応を行うので、エラー率も高くなります。エラーによって生成する点変異は、コードするタンパク質のアミノ酸変異を引き起こすので問題となります。このような人工変異を避けるには、正確度の高いポリメラーゼを用いることとPCR工程のサイクル数を少なくすることが必要です。
(ii)キメラクローン
両端に制限酵素切断部位を有するcDNAをベクターに挿入する際、制限酵素切断部位同士で連結したキメラcDNAが生成する可能性があります。
(iii)コピークローン
PCRの増幅工程や形質転換後ライブラリーの増幅を行うことによってコピークローンが生成します。完全長cDNAのコピークローンであれば問題ありませんが、短鎖cDNAの場合は問題となります。完全長かどうかの判定法の一つは、5’端の塩基配列が同じものが複数取れていることなので、短鎖cDNAクローンのコピーが存在すれば、この判定はできなくなります。
完全長cDNAライブラリーの品質の評価
表にあげた方法で作製された完全長cDNAライブラリーの品質について比較検討してみます。
(1)完全長率
転写開始点がわかっていない遺伝子の場合には、得られたcDNAがキャップ部位から始まるものかどうかわかりません。これまでは転写開始点がすでにわかっているものと同じかあるいはその近辺から始まるものを完全長cDNAとみなし、転写開始点が決められていないものについては、開始コドンから始まるORFを有するものを、完全長cDNAの候補としてカウントしてきました。各方法の完全長率を比較するには、転写開始点がわかっていて含有量の多い同じ遺伝子について完全長率を求め比較するのが妥当ですが、そのようなデータは入手できないので、異なる遺伝子集団について求めた結果を比較することにします。
(i)Okayama-Berg法
ウサギ網状赤血球のcDNAライブラリーから得た21個のα-グロビンcDNAと12個のβ-グロビンcDNAを解析した結果、10%がキャップ部位から始まる完全長cDNAクローンであり、約30〜50%が5’端の2〜3塩基目から始まるほぼ完全長cDNAクローンであることが示されました。
(ii)Pruitt法
ヒト組織球性リンパ腫細胞株U937のcDNAライブラリーから得た既知遺伝子326種のcDNAクローンのうち246クローン(75%)がORFを有していました。転写開始点がすでにわかっている遺伝子についても、ほぼ同程度の完全長率が得られています。
(iii)キメラオリゴキャッピング法
ヒト繊維肉腫細胞株HT-1080のcDNAライブラリーから得た既知遺伝子369種のcDNAクローンのうち287クローン(78%)がORFを有していました。転写開始点がすでにわかっている遺伝子についても、ほぼ同程度の完全長率が得られています。
(iv)オリゴキャッピング法
ヒト神経芽腫細胞株SK-N-MCのcDNAライブラリーから得た既知遺伝子35種のcDNAクローンのうち28クローン(80%)が転写開始点から始まる完全長cDNAでした。このライブラリーには9個のEF-1αcDNAが含まれていましたが、その中の8個(89%)が転写開始点から始まる完全長cDNAでした。さらに、ヒト胚性癌細胞株NT2のcDNAライブラリーから得た既知遺伝子67種のcDNAクローンのうち54クローン(81%)が完全長cDNAでした。
(v)キャップトラッパー法
マウス脳のcDNAライブラリー50,000ファージプラークに対して、EF-1αとGAPDHの5’端と3’端のプローブによるハイブリダイゼーションを行った結果、完全長率はそれぞれ95%と98.5%という結果を得ました。このライブラリーから任意に選んだ120クローンについて5’端の部分塩基配列を決定した結果、約94%のクローンが完全長cDNAであると判定されました。
(vi)SMART法
ヒト骨格筋のcDNAライブラリーから得た既知遺伝子84クローンのうち65クローン(77%)がORFを有していました。大規模解析を行った例としては、ヒト子宮内膜癌細胞株のcDNAライブラリーについて、サイズ分画して得られた既知遺伝子クローンの完全長率は、2~3kbpでは55.8%(362/649)、3~4kbpでは58.9%(248/421)、4~10kbpでは46.1%(170/369)でした。
(vii)ベクターキャッピング法
この方法を用いると、5’端にゲノム配列にない余分なdGが付加しているかどうかで得られたクローンが完全長かどうかを判定できます。ヒト網膜色素上皮細胞株ARPE-19のcDNAライブラリーから得た発現量が多い(>0.5%)14種の既知遺伝子455クローンのうち447個(98.2%)が転写開始点から始まる完全長cDNAでした。
以上7つの方法を比較してみると、90%以上の完全長率が得られているのは、キャップトラッパー法とベクターキャッピング法です。キャップトラッパー法は完全長cDNAを選別しているので妥当な結果ですが、ベクターキャッピング法はそのような選別工程がなくとも高い完全長率を得ています。このことは、これらの細胞内でmRNAの分解はほとんど起こっていないことを示唆しています。
Okayama-Berg法、Pruitt法、SMART法で生成する短縮cDNAは出発材料に含まれるmRNA分解産物に由来します。したがって、高品質の全RNAを使用すれば、完全長率を高めることは可能であると考えられます。一方、オリゴキャッピング法とキメラオリゴキャッピング法で生成する短縮cDNAはmRNAの処理工程で分解したmRNAに由来すると考えられます。もしmRNAの品質が高くほどんと分解していないのなら、完全長mRNAの選別の必要はなく、むしろmRNAの処理工程で分解が起こるため、かえって完全長率を下げてしまう可能性があります。
(2)複雑度
ライブラリーの複雑度を評価するには、少なくとも10,000クローン以上の部分塩基配列を決定して、発現プロフィールを求める必要があります。具体的には、mRNAの含有量とライブラリーに含まれるcDNAクローンの数が相関しているか、500bp以下の短鎖遺伝子cDNAや7kbp以上の長鎖遺伝子cDNAがどれだけ含まれているかを調べることが必要です。各方法で作製した一つのライブラリーについての詳細な検討はなされていないので比較することはできませんが、長鎖遺伝子の完全長cDNAがどれだけ取れているかが複雑度の一つの指標になると考えられます。
オリゴキャッピング法では、約100種類のヒト細胞ライブラリーから任意に選んだ115万クローンの5’端部分塩基配列を決定しましたが、6kbp以上の完全長cDNAはほとんど得られていません。キャップトラッパー法では、マウス細胞ライブラリーから選んだ144万クローンの解析で7kbp以上の長鎖完全長cDNAが18個しか採れていません。SMART法では、10種類のヒト細胞ライブラリーからサイズ分画した5万クローンの5’端部分塩基配列解析により、7.3~10kbp(クローンの実測値ではなくヒットする既知遺伝子のサイズ)のクローン568個中100個(17.6%)が完全長cDNAクローンであると推定しています。ベクターキャッピング法では、1種類のヒト培養細胞株のライブラリーから任意に選んだ24,000クローンの5’端部分塩基配列を決定したところ、7~13kbpの既知遺伝子27種のcDNAクローン62個中61個(98.4%)が完全長cDNAクローンでした。ベクターキャッピング法では、長鎖遺伝子の希少スプライシングバリアントcDNAが得られることと各種サイズのmRNAの含有量とライブラリーに含まれるcDNAクローンの数が相関していて発現量バイアスやサイズバイアスが小さいことが示されました(K08-1)。
(3)人工産物
いずれの方法でも、逆転写酵素やDNAポリメラーゼの塩基取り込みミスによって人工変異が生成する可能性はあります。特にPCR工程を含む場合は、その確率が高くなります。点変異の場合、人工変異なのか一塩基多型(SNP)なのかわかりませんが、ゲノム配列がわかっていればその区別がつきます。同じ遺伝子に由来するcDNAクローンが複数得られて、それらを比較するといくつかの違いがみられることがありますが、ほとんどの場合はSNPです。我々はcDNAの中に見出されたSNPについて幾つか論文を出しています。明らかに人工変異である例にも一度遭遇しました。巨大タンパク質であるGOLGB1のcDNAの翻訳領域にゲノム配列にない余分なAの挿入のあるクローンがとれました(3.7. Long-sized transcripts in K08-1)。ちょうどAストレッチのある部分なので、逆転写酵素の読み取りミスによるものではないかと思われます。
リンカーやアダプターを用いる場合、キメラクローンが生成することがあります。実際、リンカーを用いるGubler-Hoffman変法でキメラクローンを生成した経験があります。キャップトラッパー法でもキメラクローンができていることを見つけました(具体例)。ベクタープライマーを用いるOkayama-Berg法、Pruitt法、キメラオリゴキャッピング法、ベクターキャッピング法や、制限酵素としてSfiIを用いるオリゴキャッピング法とSMART法では、原理的にキメラクローンは生成しません。
結び
これまでトランスクリプトーム解析に用いる完全長cDNAライブラリーの作製法として実績があるのは、オリゴキャッピング法、キャップトラッパー法、SMART法、ベクターキャッピング法の4つの方法です。いずれの方法を用いても完全長cDNAクローンを取得することはできるので、どの方法を用いるかはライブラリーの利用目的に合わせて選べば良いということになります。
タンパク質を手に入れるために完全長cDNAクローンをそろえるのが目的であれば、ベクターキャッピング法が最良の選択です。ベクターキャッピング法を用いると、2μg程度の少量の全RNAから3〜4という少ない工程数で、95%以上の高い完全長率で、500bp以下の短鎖遺伝子cDNAから10kbp以上の長鎖遺伝子cDNAと0.01%以下の希少遺伝子cDNAを含む複雑度の高いライブラリーを作製できます。また、キメラクローンやコピークローンの生成も抑えられています。
高品質のmRNAの取得が難しい生物材料を用いざるを得ない時は、完全長cDNAの選別工程を含むオリゴキャッピング法やキャップトラッパー法の利用が考えられます。また、ごく微量のmRNAしか得られない場合には、ngオーダーの全RNAから完全長cDNAライブラリーを作製できるSMART法の利用が考えられます。
最近、トランスクリプトーム解析にはRNA-seqを用いることが多くなってきました。しかし、この方法では配列情報が得られても物としてのcDNAは得られません。また、遺伝子によっては、数多くのスプライシングバリアントが存在しているので、部分配列を決めただけでは、正確なスプライシングバリアントの全長配列を決めることはできません。ナノポアシーケンスによってmRNAの配列を直接読むことも可能ですが、これも得られるのは情報だけです。
塩基配列情報さえわかれば、mRNAからPCRによって必要なcDNAをクローン化できます。しかし微量しか得られない貴重なRNAサンプルの場合は、多くの遺伝子のクローンを得ることはできません。このような場合、サンプルの一部を完全長cDNAライブラリーの形で保存しておけば、これをリソースとして何度でも完全長cDNAのクローニングに使えます。
すでに多くの解析がなされている生物組織の場合も、完全長率と複雑度の高い完全長cDNAライブラリーをリソースとして保存しておけば、超長鎖遺伝子や希少遺伝子、希少スプライシングバリアント探索のために利用できます。初めてトランスクリプトーム解析を実施する生物組織の場合は、個別クローンの網羅的解析を行うことによって、発現プロフィールの解析と物としてのcDNAクローンの取得を同時に行うことができます。以上のように、リソースとして完全長cDNAライブラリーの利用価値は高いので、今後も広く活用されることを期待しています。